- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(3):326-337. doi:10.7150/ntno.20233 This issue Cite
Research Paper
TRAIL-functionalized gold nanoparticles selectively trigger apoptosis in polarized macrophages
Yen-Jang Huang1, Shan-hui Hsu1,2 ![]()
1. Institute of Polymer Science and Engineering, National Taiwan University, Taipei, Taiwan, R.O.C.
2. Institute of Cellular and System Medicine, National Health Research Institutes, Miaoli County, Taiwan, R.O.C.
Received 2017-3-25; Accepted 2017-6-15; Published 2017-7-7
Abstract

Tumor-associated macrophages (TAMs) have the same immunosuppressive effects as M2 macrophages in tumor progression and are correlated with poor-patient prognosis and survival in non-small cell lung cancer (NSCLC). Therefore, TAMs are the potential targets for cancer therapy. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of tumor necrosis factor superfamily and selectively induces cancer cell apoptosis, but not in most normal cells. Nanoparticles coated with multiple ligands can act as multivalent ligands that may actively crosslink cell surface receptors to affect downstream signals. Here, we explored nanogolds coated with TRAIL protein (nanogold-TRAIL complexes) as a potential anti-M2 macrophage drug. The structure of nanogold-TRAIL complexes comprised nanogold (3, 13, or 30 nm) as the core to crosslink multiple TRAIL for exhibition of multivalent property. Nanogold-TRAIL complexes selectively increased the cytotoxicity of TRAIL (30-fold increase in IC50) via changing O-glycosylation levels in M2-polarized macrophages. By testing the TRAIL complex efficacy on nanogold with different sizes and origins as well as on superparamagnetic iron oxide nanoparticles, we further demonstrated that the enhanced cytotoxicity by nanoparticles was dependent on size and surface properties of the nanoparticles. Meanwhile, the nanogold-TRAIL complexes remained nontoxic to M1 macrophages or normal cells. Nanogold-TRAIL complexes thus provide a novel and promising strategy for the improvement of TRAIL-based therapy.
Keywords: tumor necrosis factor-related apoptosis-inducing ligand, tumor-associated macrophages, nanogold, apoptosis, O-glycosylation.
Introduction
Non-small cell lung cancer (NSCLC) accounts for about 85% lung cancer cases throughout the world and is one of the most serious malignancy with high incidence, mortality, and poor survival [1]. The tumor microenvironment is composed of cancer cells and various stromal cells including cancer-associated fibroblasts (CAFs), endothelial cells, macrophages, and infiltrating immune cells, and has been seen a key contributor for cancer progression, epithelial-mesenchymal transition, angiogenesis, metastasis, and development of drug resistance [2, 3]. Therefore, therapies designed to target the cellular components of tumor microenvironment have started to be applied in clinical treatments [3, 4].
Macrophages are innate immune cells and have an essential role in the host defense and maintenance of tissue homeostasis [5]. The circulating monocytic precursor cells can extravasate into target tissues where they differentiate and polarize into distinct macrophage subtypes to deal with various microenvironmental challenges [3]. Each polarized macrophage exhibits a distinguishing expression profile of cytokines, enzymes, and surface markers. According to their function, polarized macrophages are generally classified into two groups: classical M1 and alternative M2 macrophages [6]. The M1 macrophages is driven by Toll-like receptor (TLR) signals and Th1 cytokine interferon-γ (IFN-γ). They are defined by the high production of pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α). Moreover, the M1 macrophages have high activities for tumor-specific antigen presentation by the increased expression of major histocompatibility complex class I (MHC-I) and class II (MHC-II) molecules. Hence, the M1 macrophages are regarded as a vital cellular component in the inflammatory response and antitumor immunity [7]. On the contrary, the Th2 cytokines interleukin-4 (IL-4) and interleukin-13 (IL-13) can induce the conversion of macrophages to the M2 phenotype which display anti-inflammatory and pro-tumorigenic activities [6].
The macrophages which are recruited by tumor cells and infiltrate into tumor tissues are commonly termed tumor-associated macrophages (TAMs). TAMs were considered with the same immunosuppressive effects as M2 macrophages in tumor progression [8]. Numerous clinical studies have reported that the TAM counts in tumors are correlated with poor-patient prognosis and survival in many cancers, and the high M1/M2 ratio of TAMs has significant association with the extended survival in cancer patients [9, 10]. Therefore, TAMs may be the potential targets for cancer therapy.
Activation of apoptosis signals is an important strategy for tumor therapies. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is one member of tumor necrosis factor (TNF) superfamily and selectively induces apoptosis in tumor cells through death receptor-mediated extrinsic pathways, but not in normal cells [11]. The trimerized TRAIL binds to the cell surface death receptor 4 (DR4) and death receptor 5 (DR5) and causes the engagement. Subsequently, Fas-associated death domain (FADD) and procaspase-8 are recruited to form the death-inducing signaling complex (DISC). DISC activates caspase-8 and cleaves procaspase-3 directly. The activated caspase-3 cleaves the downstream DNA fragmentation factor 45 (DFF45), leading to apoptotic cell death [12, 13]. Systemic administration of TRAIL reduces the mammary adenocarcinoma growth in mice without obvious toxic effects that were commonly associated with the treatment of FasL or TNF-α [14]. Therefore, TRAIL is a potential drug candidate and clinical studies are currently being conducted with recombinant human TRAIL, also called dulanermin, as a promising cancer therapy [15].
Preclinical studies revealed that approximately half of the NSCLC cell lines were resistant to the apoptosis induced by TRAIL receptor targeting agents [16]. Thus, several studies have developed TRAIL-functionalized nanoparticles for efficient activation of apoptosis [17, 18]. Here, we use nanogold bound with multiple TRAIL ligands to develop nanogold-TRAIL complexes. We expect that the multivalent property of nanogold-TRAIL complexes can selectively promote death receptor activation in M2 macrophages, but not M1 macrophages or normal cells. We demonstrate that this novel strategy may increase the M1/M2 ratio and improve the TRAIL-based therapy for NSCLC treatment.
Materials and Methods
Expression and purification of the recombinant human TRAIL protein
The recombinant bioactive TRAIL proteins (corresponding to amino acids 95-281) were expressed and purified using histidine-tagged human TRAIL complementary DNA cloned into the pRSETb plasmid [19]. The recombinant TRAIL proteins were expressed in IPTG induced Escherichia coli BL21 (DE3) cells and purified using immobilized metal-ion affinity chromatography Talon® metal affinity resins (Clontech, USA) according to the recommendations of the manufacturer. The purified TRAIL proteins were detected by immunoblotting using an anti-His tag monoclonal antibody (GeneTex, USA).
Preparation of various nanoparticles
The citrate-capped nanogolds with various diameters were purchased from Yu-Shing Biotech Co., Ltd., Taiwan. The superparamagnetic iron oxide nanoparticles (SPIO) were prepared by chemical co-precipitation [20]. FeCl2· 4H2O (8.95 g) and FeCl3· 6H2O (18.25 g) were dissolved in 150 ml distilled water. The solution was slowly added 50 ml NaOH with stirring for 30 min, followed by a colour change from light brown to black. Fe3O4 NPs formed as the precipitate and were then washed three times in distilled water by centrifugation. Sodium oleate was added to the Fe3O4 solution with vigorous stirring for 30 min and then the excess surfactant was removed via dialysis [20]. The nanogolds with 30 nm diameter (GNT30, Gold NanoTech Co., Ltd., Taiwan) were produced via physical manufacturing without any surface modifiers or stabilizers [21].
Adsorption of TRAIL on various nanogolds
The recombinant TRAIL proteins were dialyzed in phosphate buffer (10 mM) by a centrifugal ultrafilter (30 kDa MWCO, Millipore, USA). To prepare the nanogold-TRAIL complexes, TRAIL proteins (50 μg) were added to 1 ml of different nanogold solutions (Evernew, Taiwan, 200 μg/ml of nanogolds with various sizes) and were stirred for 10 min, and then the solution was centrifuged at 12,000×g (at 4ºC) for 30 min to remove unbound excess TRAIL proteins. The pellet was resuspended in 10 mM phosphate buffer. The protein concentration in the supernatant was measured using a micro BCA assay kit (G-biosciences). The amount of TRAIL proteins bound to the surface of nanogolds was calculated using the following formula: protein bound to nanogolds = total protein added - protein in the supernatant solution [22].
Characterization of the nanogold-TRAIL complexes
The nanogolds were diluted to 50 μg/ml, cast on a mesh copper grid, and dried out at the fume hood. The sizes and shapes of various nanogolds were determined by transmission electron microscopy (TEM, JEM-1200EX II, JEOL, Japan). The UV-visible spectroscopy, dynamic light scattering (DLS), and zeta potential were used to identify the features of nanogolds coated with TRAIL proteins. The absorption spectra of different nanogolds and nanogold-TRAIL complexes were recorded from 400-700 nm with an interval of 1 nm by UV-visible spectroscopy (SpectraMax® M5, Molecular Devices, USA). The hydrodynamic sizes of all nanogolds and nanogold-TRAIL complexes were measured by DLS using a cumulant method. The zeta potential values were determined by the electrophoretic light scattering measurements. Each measurement of size and zeta potential was performed at 25°C by a submicrometer particle analyzer (Delsa Nano Analyzer, Beckman Coulter). The average value was represented by the arithmetic mean (± standard deviation) from three different samples.
Cell culture of M0 macrophages and induction of M1 and M2 macrophages
Human monocytic THP-1 cells were grown in continuous culture in Roswell Park Memorial Institute medium (RPMI 1640, Gibco) supplemented with 10% heat inactivated fetal bovine serum (FBS, Gibco) and 10 mg/l penicillin-streptomycin. Cells were maintained in a humidified incubator at 37ºC and 5% CO2, and subcultured twice per week. THP-1 monocytes were differentiated into macrophages by incubation with 200 ng/ml phorbol 12-myristate 13-acetate (PMA, caymanchem) for 48 h. For M1 polarization, macrophages were incubated with 20 ng/ml of human interferon gamma (IFN-γ, peprotech) and 200 ng/ml of lipopolysaccharide (LPS, Sigma). For M2 polarization, macrophages were incubated with 20 ng/ml of interleukin 4 (IL-4, peprotech) and 20 ng/ml of interleukin 13 (IL-13, peprotech).
L929 murine fibroblast cell line, and bovine endothelial cells (BECs) were used to evaluate the selective cytotoxicity of nanogold-TRAIL complexes. L929 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM, Gibco) high glucose with 10% FBS (Gibco) and 10 mg/l penicillin-streptomycin. BECs were cultured in DMEM low glucose (Gibco) containing 10% FBS (Gibco) and 10 mg/l penicillin-streptomycin.
Cell viability by the Cell Counting Kit-8 (CCK-8) assay
Cells (2×104) were plated in 96-well plates for 24 h and then incubated with different concentrations of TRAIL or nanogold-TRAIL in RPMI-1640 medium. Following 24 h incubation, the cell viability was determined by the CCK-8 assay (Sigma). Cell viability was determined as percent of viable cells relative to an untreated control. IC50 was defined as the protein concentration causing a 50% decrease of absorbance at 450 nm compared with the untreated control. The absorbance was measured at 450 nm using a microplate reader (SpectraMax® M5, Molecular Devices, USA).
Cell vitality and apoptosis by fluorescence staining
The cellular health of M2 macrophages was evaluated by VitBright-48™ (VB-48, Chemometec) and propidium iodide (PI, Chemometec). VB-48™ was used to determinate the level of thiols, such as reduced glutathione (GSH), which is involved in the progression of cell death. M2 macrophages (1 × 106) were incubated with TRAIL or nanogold-TRAIL for 4 h and washed with PBS. Cells were collected by trypsinization and centrifugation. Cells were stained with VB-48™ and PI for 5 min. For cell cycle analysis, M2 macrophages (1 × 106) were incubated with TRAIL or nanogold-TRAIL for 8 h. Cells were stained with 4',6-diamidino-2-phenylindole (DAPI, chemometec) at 37°C for 5 min. The cellular fluorescence is analyzed and quantified by the NucleoCounter® NC-3000™.
Analysis of CD markers and O-glycosylation expression by flow cytometry
THP-1 monocytes were seeded in 6-well plates at 5×105 cells/well and were treated with PMA. After incubation with LPS/IFN-γ or IL-4/IL-13 for 48 h, cells were washed with PBS and detached with the culture medium. Cell suspension was centrifuged for 5 min at 1000 rpm and the pellet resuspended with 1 ml of PBS. This washing step was performed twice. The cell pellets were resuspended with 4 % paraformaldehyde in PBS and incubated for 15 min at 4 °C. After the washing step was performed twice by centrifugation in PBS, the cells were incubated with primary anti-CD86 (Abcam), anti-CD206 (Abcam), anti-O-linked N-acetylglucosamine (O-GlcNAc, Genetex) antibodies, and the control isotype (Genetex) in phosphate-buffered saline containing 0.1% (v/v) Tween-20 (PBST) and 2% bovine serum albumin (BSA) and incubated for 2 h at 4°C. After the washing step was performed twice by centrifugation in PBST, the cells were incubated with secondary antibodies with Alexa Fluor® 488 (abcam) corresponding to each primary antibody in PBST with 2% BSA and incubated for 2 h at 4 °C. After the washing step was performed twice by centrifugation in PBST, the cells were resuspended in 1 ml of PBST. Cells were then analyzed the fluorescence intensity by flow cytometry (FACScalibur, BD Biosciences).
Cytokine profiling by the enzyme-linked immunosorbent assay (ELISA)
THP-1 monocytes were seeded in 6-well plates at 5×105 cells/well and were treated with PMA. After incubation with LPS/IFN-γ or IL-4/IL-13 for 48 h, secreted cytokines were analyzed in cell culture medium by ELISAs (R&D Systems).
Quantitative real time reverse transcription-polymerase chain reaction (qRT-PCR) analysis
The levels of mRNA expression for macrophage markers, death receptors and O-glycosylation enzymes were analyzed by qRT-PCR. Total cellular RNA were isolated by Trizol® reagent (Invitrogen). The extracted RNA was reverse-transcribed into cDNA by the RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, St. Leon-Rot, Germany). qRT-PCR was performed in a StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific Inc, USA) by the DyNAmo Flash SYBR Green qPCR Kit (Finnzymes Oy, Espoo, Finland). Products were detected using primers for CD36, CD68, DR4, DR5, Polypeptide N-Acetylgalactosaminyltransferase 14 (GALNT14), fucosyltransferase 6 (FUT6), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The levels of gene expression were normalized to those of GAPDH. The normalized values were shown as the relative ratio compared to that in the mock group. The primer sequences used in this study are listed in Supplementary Table S1.
Caspase 8 and caspase 3 activity assay
The colorimetric caspase 8 and caspase 3 assay (BioVision, USA) was performed according to the manufacturer's instructions. Briefly, after 6 h treatment with TRAIL or nanogold-TRAIL, cells were lysed and centrifuged at 12,000 g (30 min, 4ºC). The equal amount of protein (50 µg) of cell lysates for each group was reacted with the mixture containing substrates of caspase 8 or caspase 3 for 2 h at 37°C. The absorbance was measured at 405 nm by the microplate reader (SpectraMax® M5, Molecular Devices, USA).
Statistical analysis
Data were shown as the mean ± standard deviation. Each type of experiment was executed independently for multiple times to assure reproducibility. Typical data were displayed in this study. Statistical differences among the experimental groups were evaluated by analysis of variance followed by one-way ANOVA. The p-value less than 0.05 was considered significant.
Results
Characterization of polarized macrophages
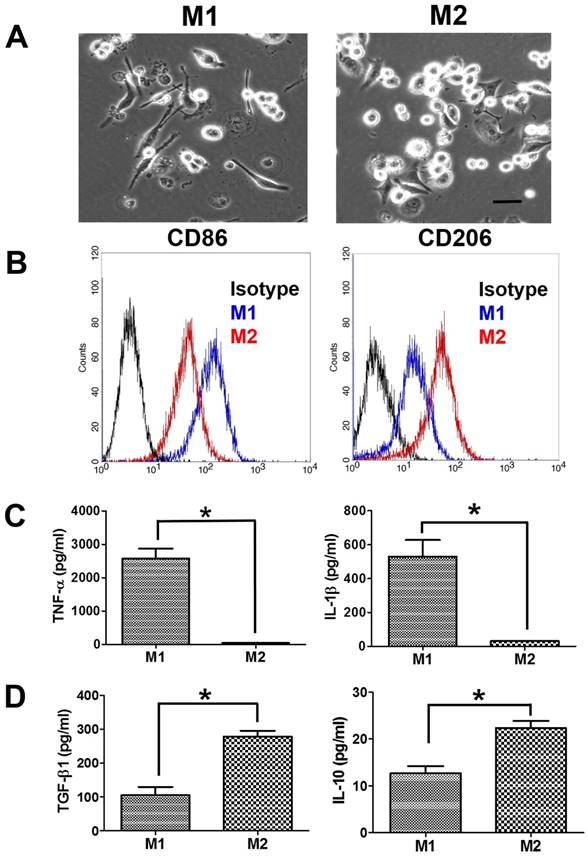
The suspended human THP-1 monocytes were differentiated into macrophages by incubation with PMA. The differentiated M0 macrophages became adherent and had the spindle or circular morphology. M0 macrophages also showed the increase in the gene expression of macrophage markers (CD36 and CD68) compared to THP-1 monocytes (Supplementary Figure S1). M0 macrophages further treated with LPS/IFN-γ for M1 polarization demonstrated a dendritic-like morphology with large filopodia, whereas M2 macrophages remained the spindle or round shape (Figure 1A). Next, we assessed the macrophage polarization by flow cytometric analysis of CD surface markers. M1 macrophages had greater CD86 expression than M2 macrophages, and M2 macrophages had greater CD206 expression than M1 macrophages (Figure 1B). Besides, M1 macrophages displayed more pro-inflammatory cytokines, TNF-α and IL-1β, compared to M2 macrophages (Figure 1C). Meanwhile, the protein expression levels of immunosuppressive cytokines, IL-10 and TGF-β, were greater in M2 macrophages than in M1 macrophages (Figure 1D).
Characterization of polarized macrophages. (A) The morphologies of polarized macrophages after incubation with LPS/IFN-γ or IL-4/IL-13 for 48 h. The scale bar represents 20 μm. (B) The protein expressions of CD markers for M1 macrophages or M2 macrophages after incubation with LPS/IFN-γ or IL-4/IL-13 for 48 h. (C, D) The protein expressions of cytokines for M1 macrophages or M2 macrophages after incubation with LPS/IFN-γ or IL-4/IL-13 for 48 h. *p < 0.05, among the indicated groups.
O-glycosylation controls polarized macrophage sensitivity to TRAIL
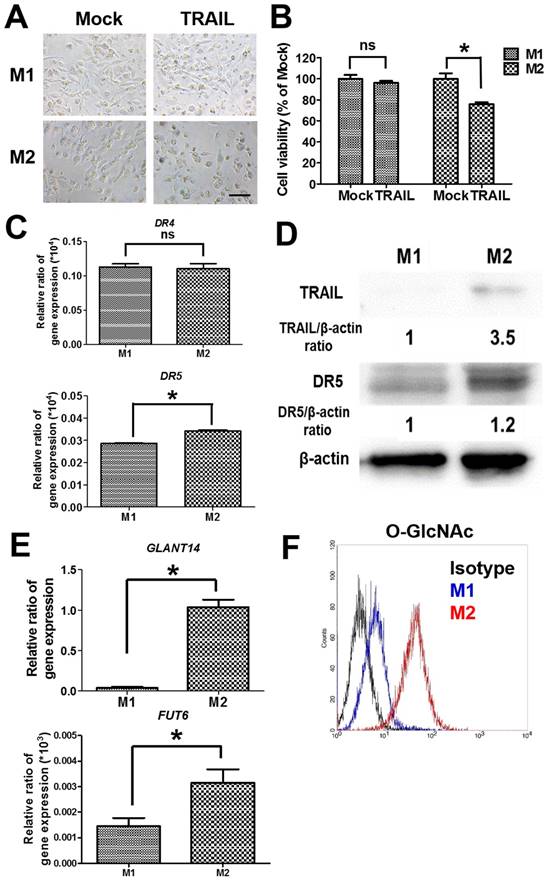
To examine the cytotoxicity of TRAIL in polarized macrophages, both M1 and M2 macrophages were treated with TRAIL, respectively. After the incubation with TRAIL protein, M1 macrophages kept a dendritic morphology (Figure 2A) and had no obvious change in viability (Figure 2B). Meanwhile, many M2 macrophages showed shrinkage in shape and exhibited lower viability after TRAIL treatment (Figure 2A and B). Since death receptor mediated the TRAIL signaling, we evaluated the expressions of death receptors DR4 and DR5 in M1 and M2 macrophages. The gene expressions of DR4 were the same in both M1 and M2 macrophages, but the DR5 gene expression in M2 macrophages was a little higher than that in M1 macrophages (Figure 2C). Consistent with the result of DR4, the expression level of DR5 protein in M2 macrophages was also slightly higher than that in M1 macrophages (Figure 2D). Nevertheless, the affinity of TRAIL in M2 macrophages was 3.5 folds greater than that in M1 macrophages (Figure 2D). Since the O-glycosylation level of cells modulated the cell sensitivity to TRAIL proteins, we compared O-glycosylation level between M1 and M2 macrophages. The mRNA expressions of GALNT14 and FUT6 genes, encoding the O-glycan processing enzymes, were markedly greater in TRAIL sensitive M2 macrophages versus TRAIL resistant M1 macrophages (Figure 2E). By flow cytometric analysis of antibodies directed against glycan structures, we further confirmed that M2 had a greater O-glycosylation level than M1 macrophages (Figure 2F). These results indicate that the level of O-glycosylation may facilitate the sensitivity of TRAIL in different polarized macrophages.
The cytotoxicity and affinity of TRAIL in polarized M1 and M2 macrophages by O-glycosylation. (A) Cell morphology of polarized M1 and M2 macrophages after TRAIL (250 ng/ml) treatment for 24 h. Scale bar = 50 μm. Mock indicated the untreated group. (B) The viability of polarized M1 and M2 macrophages after TRAIL (250 ng/ml) treatment for 24 h. (C) The mRNA expressions of DR4 and DR5 for M1 and M2 macrophages. (D) The affinity of M1 and M2 macrophages to TRAIL molecule after incubation with TRAIL (250 ng/ml) for 2 h. The relative protein expression was quantified by the software ImageJ. (E) The gene expression of GLANT14 and FUT6 in M1 and M2 macrophages. (F) The levels of O-GlcNAc proteins for M1 macrophages or M2 macrophages by flow cytometry. Each bar represents the means of three determinations ± SD. *p < 0.05, among the indicated groups.
Synthesis and basic properties of nanogold-TRAIL complexes
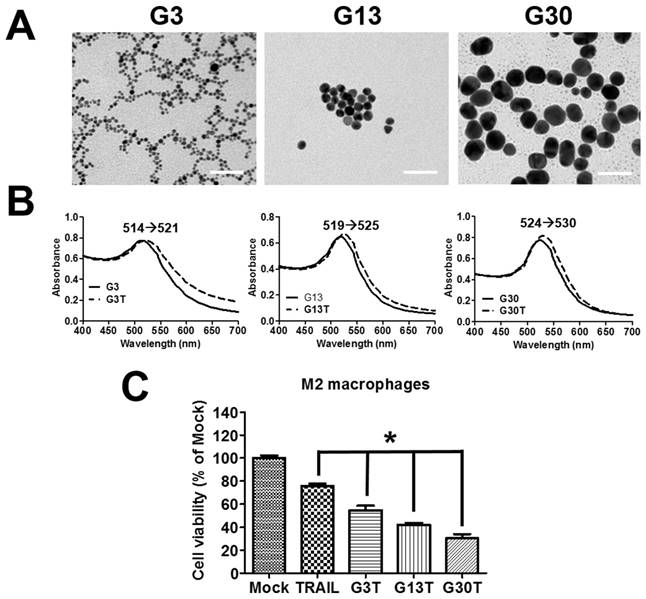
The molecular weight of purified recombinant human TRAIL was about 26 kDa and could be recognized by anti-His antibody (Supplementary Figure S2A and B). Furthermore, it exhibited obvious cytotoxicity to H460 cells, which were TRAIL-sensitive cells (Supplementary Figure S2C). Hence, the purified recombinant human TRAIL had the right molecular weight and bioactivity. The regulation by ligand-coated nanoparticle on the process of membrane receptor signals is strongly dependent on the nanoparticle size [23]. Here, the recombinant human TRAIL proteins conjugated onto citrate-coated nanogold surface with various size (3, 13, and 30 nm) were prepared to generate multivalent properties of nanoparticle-ligand complexes (G3T, G13T, and G30T). TEM images showed that 3 nanogolds of each size had spherical shape with a rather uniform size (Figure 3A). The diameter and zeta potential of these nanoparticles determined by light scattering are shown in Supplementary Table S2. The hydrodynamic sizes of various nanogolds were larger than those obtained from TEM images. Since nanogolds were conjugated with TRAIL protein, all nanogold-TRAIL complexes had larger hydrodynamic diameters and less negative zeta potential than uncoated nanogolds. Moreover, G3, G13, and G30 had strong visible light absorption with peaks at 514, 519, and 524 nm in the spectra, which were shifted to 521, 525, and 530 nm after TRAIL conjugation, respectively (Figure 3B). The absorbed amount of TRAIL on various nanogolds (1 μg) were about 0.25 μg in G3, 0.13 μg in G13, and 0.1 μg in G30, respectively. We further examined the cytotoxicity of various nanogold-TRAIL complexes in M2 macrophages. As shown in Figure 3C, the viability of M2 macrophages was reduced to 75-80% after free TRAIL treatment. The cell viability was decreased more remarkably by the treatment of nanogold-TRAIL complexes vs. the free TRAIL. The nanogold-TRAIL (G3T, G13T, and G30T) treated M2 macrophages only demonstrated 30-55% viability. Furthermore, G30T caused the greatest reduction in cell viability among the nanogold-TRAIL complexes (Figure 3C). These results indicated that the cytotoxic effect of TRAIL on M2 macrophages by TRAIL was augmented after TRAIL was bound to nanogolds. Moreover, the cytotoxicity was dependent on the size of the nanogold-TRAIL complexes, with nanogolds of 30 nm demonstrating the most significant effect.
The properties of nanogold-TRAIL complexes. (A) TEM images of nanogolds with different sizes. Scale bar = 50 nm. (B) Spectroscopic detection of the absorption peak for different sizes of nanogolds before and after TRAIL binding. (C) M2 macrophages were treated with different nanogold-TRAIL complexes respectively for 24 h. The TRAIL concentration was 500 ng/ml. Each bar represents the means of three determinations ± SD. *p < 0.05, among the indicated groups.
Nanogold-TRAIL complexes exhibit selective cytotoxicity to polarized macrophages
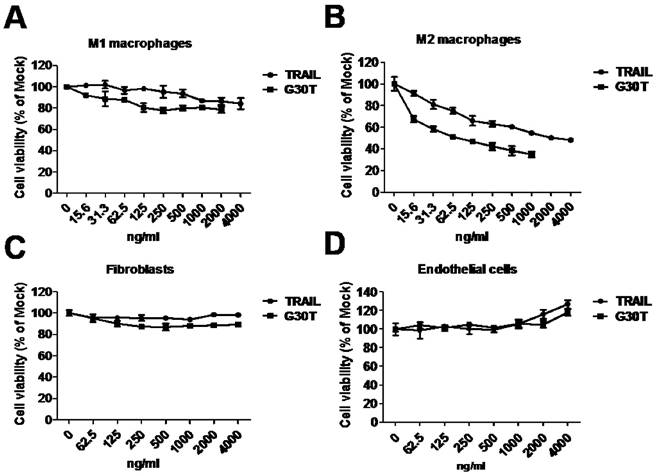
To evaluate the sensitivity of polarized macrophages to nanogold-TRAIL induced cytotoxicity, M1 and M2 macrophages were incubated with various concentrations of TRAIL and G30T. As shown in Figure 4A, the cell viability of M1 macrophages was only slightly decreased after TRAIL or G30T treatment of various concentrations. On the other hand, G30T treated M2 macrophages showed much lower cell viability with the same protein dosage of TRAIL. The IC50 values for TRAIL and G30T were 2232 ng/ml and 75 ng/ml, respectively. In another word, the G30T showed about 30 fold increase in cytotoxicity for M2 macrophages compared to TRAIL (Figure 4B). Furthermore, nanogolds of various sizes demonstrated no obvious cytotoxicity in M2 macrophages (Supplementary Figure S3). To verify the safety, we also evaluated the sensitivity of normal cells to nanogold-TRAIL induced cytotoxicity. For both fibroblasts and endothelial cells, the cell viabilities were not significantly reduced by TRAIL and G30T at various doses (Figure 4C and D). Moreover, the viability of endothelial cells slightly increased at high doses of TRAIL or G30T treatments. These data suggested that TRAIL bound to the nanogold surface resulted in an obvious increase of cytotoxicity in M2 macrophages while remained the selectivity and nontoxicity in M1 macrophages and normal cells.
The cytotoxicity of G30T in various types of cells. (A, B) The viability of M1 and M2 macrophages after treatment with various concentrations of TRAIL (15.6 to 4000 ng/ml) or G30T (15.6 to 2000 ng/ml) for 24 h. The corresponding gold amount in the latter was 0.156 to 20 μg/ml. (C, D) The viability of fibroblasts and endothelial cells after treatment with various concentrations of TRAIL or G30T (62.5 to 4000 ng/ml) for 24 h. The corresponding gold amount in the latter was 0.625 to 20 μg/ml.
Nanogold-TRAIL complexes induce apoptosis via a caspase dependent pathway in M2 macrophages
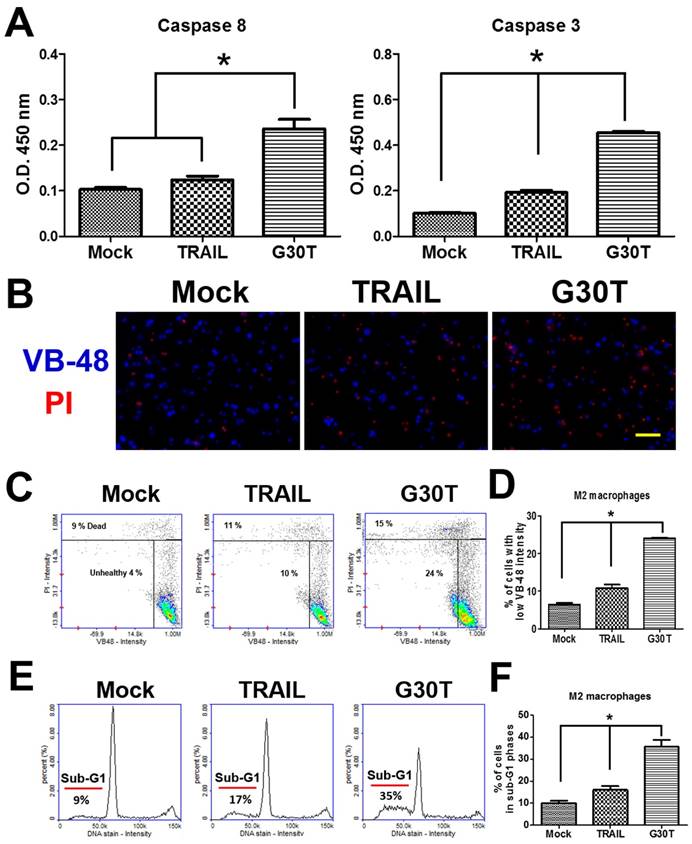
We investigated the signal pathways trigged by G30T in M2 macrophages. As shown in Figure 5A, G30T induced higher activities of caspase-8 and caspase-3 than TRAIL. Since high levels of reactive oxygen species (ROS) are associated with apoptosis, we also compared the glutathione (GSH) level by the VB-48™ intensity of M2 macrophages after TRAIL and G30T treatments. Following the G30T treatment, M2 macrophages exhibited significantly less fluorescence intensity of VB-48™ and more fluorescence intensity of PI, indicating that G30T induced more intracellular ROS and cell death (Figure 5B-D). Consistently, M2 macrophages exhibited more sub-G1 phase populations in the group of G30T treatment vs. the group of TRAIL treatment (Figure 5E and F). These studies demonstrated that the nanogold-TRAIL complexes caused stronger cytotoxicity via inducing apoptosis signals in M2 macrophages.
The nanogold-TRAIL complexes induced apoptotic signals in M2 macrophages. (A) M2 macrophages treated with the same dosage of TRAIL (250 ng/ml) or G30T were subjected to the caspase activity assay. Mock indicated the untreated group. (B) The immunofluorescence of VB-48 (live stain) and PI (dead stain) in M2 macrophages after the treatment by TRAIL or G30T for 4 h. Scale bar = 50 μm. (C) Plots of VB-48 intensity versus PI intensity in M2 macrophages after the treatment by TRAIL or G30T treatment for 4 h. (D) The average population percentage of cells with low VB-48 staining intensity (unhealthy cells). (E) Quantification of DNA for M2 macrophages after the treatment by TRAIL or G30T treatment for 6 h. (F) The average percentage of cell population in sub-G1 phases. Each bar represents the means of three determinations ± SD. *p < 0.05, among the indicated groups.
Nanoparticle enhanced TRAIL cytotoxicity is dependent on nanoparticle properties
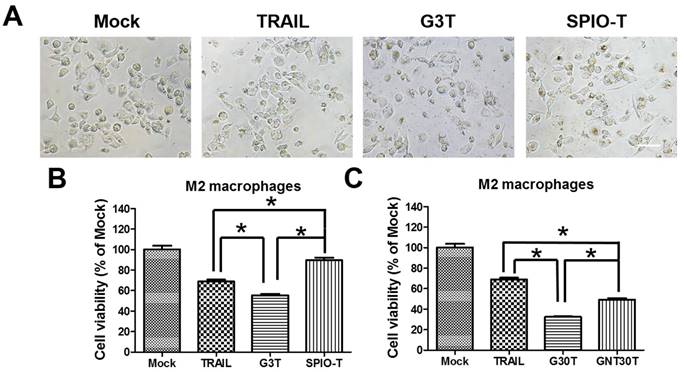
Since the nanoparticle-ligand interaction may interfere with the original activity of ligands, we further investigated the activity of TRAIL cytotoxicity after conjugation with different nanoparticles. We used superparamagnetic iron oxide nanoparticles (5 nm, SPIO) and physically manufactured nanogolds (30 nm, GNT30) to form TRAIL conjugation, namely SPIO-T and GNT30T, respectively. The amounts of adsorbed TRAIL on SPIO and GNT30 were 0.2 and 0.08 μg, respectively. As shown in Figure 6A, G3T (nanogold core with a size close to SPIO) induced more morphological shrinkage of M2 macrophages than SPIO-T or TRAIL alone. The cytotoxicity of TRAIL was enhanced after conjugation with G3 but not with SPIO of a similar size (Figure 6B). Moreover, the cytotoxic effect of G30T on M2 macrophages was more pronounced than that of GNT30T with a similar nanogold core size. Meanwhile, GNT30 had slight cytotoxicity in M2 macrophages at high concentrations, but SPIO did not (Supplementary Figure S4). These data suggested that the biological effects of TRAIL conjugated on nanoparticles of different characteristics may be distinct.
The enhanced TRAIL cytotoxicity was dependent on the choice of core nanoparticles for conjugation. (A) The cell morphology of M2 macrophages after TRAIL, SPIO-T, and G3T treatment (250 ng/ml) for 24 h. Scale bar = 50 μm. (B) The relative viability of M2 macrophages after TRAIL, SPIO-T, and G3T treatment (250 ng/ml) for 24 h. (C) The relative viability of M2 macrophages after TRAIL, G30T, and GNT30T treatment (250 ng/ml) for 24 h. Each bar represents the means of three determinations ± SD. *p < 0.05, among the indicated groups.
Discussion
Inflammatory responses play diverse roles at different steps of tumor progression, such as initiation, promotion, malignant conversion, invasion, and metastasis and have been seen as the seventh hallmark of cancer [24, 25]. TAMs have a phenotype similar to M2-polarized macrophages and are key mediators of the link between inflammation and cancer [26]. It has been reported that patients with the high infiltration intensity of M2-polarized TAMs have the negative correlation with tumor progression and poor patient outcome [27]. As M2-polarized TAMs plays an important role in the prognosis of tumor, targeting M2-polarized TAMs also means a novel direction for the therapeutic strategies. Results from our current study showed an important example for TRAIL modulated apoptosis by O-glycosylation. Our data revealed that M2 macrophages had greater levels of expression of specific O-glycosyltransferases and O-GlcNAc glycosylated proteins than M1 macrophages, and in literature the glycosylation levels were significantly correlated with the sensitivity to the TRAIL in some cancer cells [28]. Consistent with these results, we observed that M2 macrophages had higher affinity and more sensitivity to TRAIL than M1 macrophages. We further used benzyl-2-acetamido-deoxy-α-D-galactopyranoside (benzyl-α-GalNAc) to inhibit the O-glycosylation level of M2 macrophages. The cell viability of M2 macrophages was rescued with G30T treatment after benzylGalNAc incubation (Supplementary Figure S5). Hence, we considered that the O-glycosylation was correlated with TRAIL-nanogolds induced cell death. These findings suggested that targeting the O-glycosylation level of M2 macrophages may help cancer patients to respond to TRAIL-based therapy.
DISC formation plays an important role to initiate apoptotic signals through death receptor aggregation. Nanoparticles with multivalent properties by coating targeting molecules play a role in vector delivery to increase payload influx and imaging detection [22]. Furthermore, nanoparticles can be conjugated to ligands to regulate receptor distribution and downstream cell signals [22, 29]. In this work, we applied nanogolds coated with TRAIL as a new strategy to improve TRAIL-based therapy. In addition to the enhanced cytotoxicity to tumor cells (Supplementary Figure S6), which was consistent with literature [17, 18] using lipid nanoparticles, we were particularly interested in the M2 macrophage toxicity. When TRAIL was conjugated to nanogolds, enhanced cytotoxicity was observed in M2 macrophages, but not in M1 macrophages or normal cells. The cell viability of endothelial cells even slightly increased after TRAIL or G30T treatment at higher concentrations. The increased cell viability may probably be ascribed to the activation of the Akt and ERK pathways in endothelial cells, which was reported to be promoted by TRAIL [30]. Meanwhile, higher levels of apoptotic signals were induced in M2 macrophages by the nanogold-TRAIL complexes. Moreover, we observed that nanogold-TRAIL complexes promoted the activations of caspase-8 and caspase-3 and increased more cell populations in the sub-G1 phase than TRAIL. These phenomena may be attributed to the multivalent property of nanogold-TRAIL complexes, by which it could increase the binding affinity of TRAIL and death receptor activation in M2 macrophages. In fact, normal cells and M1 macrophages having rather lower O-glycosylation and affinity to TRAIL were resistant to the nanogold-TRAIL complex-induced cell death. We further analyzed the gene expressions of TRAIL-R3 and TRAIL-R4, which were decoy receptors of TRAIL. The gene expressions of TRAIL-R3 and TRAIL-R4 were equal in M1 and M2 macrophages (Supplementary Figure S7). Since the nuclear localization of DR5 reduces the TRAIL-induced cell death of human tumor cells [31], the mechanism of TRAIL sensitivity for M1 and M2 macrophages requires further investigation.
Nanoparticle-protein conjugation has various applications in sensing, imaging, delivery, catalysis, and therapy by protein structure and activity [32]. Therefore, the characteristics of the nanoparticle-protein interface, which control the protein orientation on the nanoparticle, is of great importance. In this study, we used citrate-capped nanogolds of different sizes (3-30 nm) to conjugate TRAIL proteins. The enhanced cytotoxic effects were more pronounced for nanogold of 30 nm diameter. Literature has shown that engineered nanoparticles of well-defined sizes can selectively induce membrane receptor distribution and alters the downstream signaling [23]. Since death receptor clustering on cell membrane is a key factor to promote the DISC activation and apoptosis pathways, nanogold-TRAIL complexes having multiple binding elements with death receptors may induce more intensive death receptor aggregation in M2 macrophages than TRAIL alone. Meanwhile, other surface properties are critically important. After conjugation with SPIO or nanogold prepared from a different process, the nanoparticle-TRAIL complex was not as effective. In particular, SPIO-TRAIL complex even reduced the cytotoxicity of TRAIL. These results may be associated with distinct protein configuration on the nanoparticles. Judging from our data, it appeared that the conjugated complex with less negative surface charge may be more bioactive. As conjugation often affects the protein structure and bioactivity, the TRAIL structure on different nanoparticles may be the dominant factor for cell signaling and deserve further investigations.
DISC formation is important to transmit TRAIL death signals. However, the above mechanism failed to trigger TRAIL-DISC-initiated cell death signals in TRAIL-resistant NSCLC cells [33]. Here, we observed that TRAIL-nanogold conjugates successfully induced cytotoxicity in TRAIL-resistant A549 lung cancer cells. Since death receptor clustering on cell membrane is a key factor to promote DISC activation, nanogold-TRAIL complexes having multiple binding elements with death receptors may induce more intensive death receptor aggregation than TRAIL in TRAIL-resistant cells, leading to enhanced cytotoxicity [17, 18]. Combining the effects on tumor cells and TAMs, the nanogold-TRAIL complexes may have a significant impact on the conceptual designs of developing new nanomedicines for cancer treatment, in particular for applications in ligand-based cancer therapy.
Conclusion
Nanogolds conjugated with multiple ligands (i.e. nanogold-TRAIL complexes) were utilized in this study to modulate the activation of membrane receptor (i.e. TRAIL receptor) to affect downstream cell signals of tumor-associated M2-polarized macrophages. The structure of nanogold-TRAIL complexes comprised nanogolds as the core to crosslink multiple TRAIL for exhibition of multivalent property. Using this approach, TRAIL signals could be selectively enhanced to increase the apoptosis of M2 macrophages by high O-glycosylation levels. The multivalent nanogold (30 nm)-TRAIL complexes induced significant cytotoxicity (30 folds) in M2 macrophages compared to TRAIL, via caspase-dependent pathways. This new finding will have a significant impact on developing new nanomedicines for TAM-based cancer treatment in particular for applications in TRAIL- or other ligand-based cancer therapy.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research was supported by the Program for Stem Cell and Regenerative Medicine Frontier Research (MOST103-2321-B-002-100) sponsored by the Ministry of Science and Technology and the Aim for the Top University Plan (105R4000) supported by Ministry of Education, Taiwan. We are grateful to Dr. Shiu-Huey Chou for her assistance with THP-1 monocyte cell lines, Dr. Ping-Ning Hsu for his assistance with human TRAIL plasmid, and Dr. Yuh-Shan Jou for his assistance with H460 and A549 cells.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Farhat FS, Houhou W. Targeted therapies in non-small cell lung carcinoma: what have we achieved so far? Ther Adv Med Oncol. 2013;5:249-70
2. Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA targeting to modulate tumor microenvironment. Front Oncol. 2016;6:3
3. Tsai MJ, Chang WA, Huang MS, Kuo PL. Tumor microenvironment: a new treatment target for cancer. ISRN Biochem. 2014;2014:351959
4. Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem. 2013;59:85-93
5. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593-604
6. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889-96
7. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6:1670-90
8. Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098
9. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315-22
10. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A. et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014;7:19
11. Refaat A, Abd-Rabou A, Reda A. TRAIL combinations: The new 'trail' for cancer therapy (Review). Oncol Lett. 2014;7:1327-32
12. Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139-44
13. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780-8
14. Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122-33
15. Soria JC, Smit E, Khayat D, Besse B, Yang X, Hsu CP. et al. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J Clin Oncol. 2010;28:1527-33
16. Stegehuis JH, de Wilt LH, de Vries EG, Groen HJ, de Jong S, Kruyt FA. TRAIL receptor targeting therapies for non-small cell lung cancer: current status and perspectives. Drug Resist Updat. 2010;13:2-15
17. Seifert O, Pollak N, Nusser A, Steiniger F, Ruger R, Pfizenmaier K. et al. Immuno-LipoTRAIL: Targeted delivery of TRAIL-functionalized liposomal nanoparticles. Bioconjug Chem. 2014;25:879-87
18. De Miguel D, Gallego-Lleyda A, Ayuso JM, Erviti-Ardanaz S, Pazo-Cid R, del Agua C. et al. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology. 2016;27:185101
19. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673-82
20. Hsu Sh, Ho TT, Tseng TC. Nanoparticle uptake and gene transfer efficiency for MSCs on chitosan and chitosan-hyaluronan substrates. Biomaterials. 2012;33:3639-50
21. Yen HJ, Hsu Sh, Tsai CL. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5:1553-61
22. Huang YJ, Shiau AL, Chen SY, Chen YL, Wang CR, Tsai CY. et al. Multivalent structure of galectin-1-nanogold complex serves as potential therapeutics for rheumatoid arthritis by enhancing receptor clustering. Eur Cell Mater. 2012;23:170-81 discussion 81
23. Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145-50
24. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
25. Mantovani A. Cancer: Inflaming metastasis. Nature. 2009;457:36-7
26. Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22:289-97
27. Niino D, Komohara Y, Murayama T, Aoki R, Kimura Y, Hashikawa K. et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL). Pathol Int. 2010;60:278-83
28. Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K. et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070-7
29. Shokeen M, Pressly ED, Hagooly A, Zheleznyak A, Ramos N, Fiamengo AL. et al. Evaluation of multivalent, functional polymeric nanoparticles for imaging applications. ACS nano. 2011;5:738-47
30. Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D. et al. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation. 2003;107:2250-56
31. Kojima Y, Nakayama M, Nishina T, Nakano H, Koyanagi M, Takeda K. et al. Importin β1 protein mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J Biol Chem. 2011;286:43383-93
32. Aubin-Tam ME, Hamad-Schifferli K. Structure and function of nanoparticle-protein conjugates. Biomed Mater. 2008;3:034001
33. Song JH, Tse MC, Bellail A, Phuphanich S, Khuri F, Kneteman NM. et al. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non-small cell lung carcinoma cells. Cancer Res. 2007;67:6946-55
Author contact
![]() Corresponding author: Institute of Polymer Science and Engineering, National Taiwan University, No. 1, Sec., Roosevelt Road, Taipei 10617, Taiwan, R.O.C., Phone: (886) 2-33665313, Fax: (886) 2-33665237; E-mail: shhsuedu.tw
Corresponding author: Institute of Polymer Science and Engineering, National Taiwan University, No. 1, Sec., Roosevelt Road, Taipei 10617, Taiwan, R.O.C., Phone: (886) 2-33665313, Fax: (886) 2-33665237; E-mail: shhsuedu.tw