- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Abbreviations
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2022; 6(4):388-399. doi:10.7150/ntno.71906 This issue Cite
Research Paper
Covalent core-radiolabeling of polymeric micelles with 125I/211At for theranostic radiotherapy
Emanuel Sporer1, Christian B. M. Poulie2, Tom Bäck3, Sture Lindegren3, Holger Jensen4, Paul J. Kempen1,5, Andreas Kjaer4,6, Matthias M. Herth2,4 ![]() , Andreas I. Jensen1
, Andreas I. Jensen1 ![]()
1. Center for Nanomedicine and Theranostics (The Hevesy Laboratory), DTU Health Technology, Technical University of Denmark (DTU), Ørsteds Plads 345C, 2800 Lyngby, Denmark
2. Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark
3. Departments of Radiation Physics, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, Gula Stråket 2b, 41345 Gothenburg, Sweden
4. Department of Clinical Physiology, Nuclear Medicine & PET, Rigshospitalet, Blegdamsvej 9, 2100 Copenhagen, Denmark
5. National Centre for Nano Fabrication and Characterization, Technical University of Denmark (DTU), Oersteds Plads—Building 347, 2800 Lyngby, Denmark
6. Cluster for Molecular Imaging, Department of Biomedical Sciences, University of Copenhagen, Blegdamsvej 3, 2100 Copenhagen, Denmark
Received 2022-2-14; Accepted 2022-7-7; Published 2022-7-18
Abstract

Astatine-211 (211At) is one of the most promising α-emitters for targeted alpha therapy, especially of cancer metastases. However, the lack of a stable isotope, frequent in vivo deastatination, and limited radiochemical knowledge makes it challenging to apply. Here, we report a new strategy for radiolabeling the lipophilic core of polymeric micelles (PMs) with covalently bound 211At. The PMs were radiolabeled via either an indirect synthon-based method or directly on the amphipathic block copolymer. The radiochemistry was optimized with iodine-125 (125I) and then adapted for 211At, enabling the use of both elements as a potential theranostic pair. PMs that were core-radiolabeled with both 125I or 211At were prepared and characterized, based on a PEG(5k)-PLGA(10k) co-polymer. The stability of the radiolabeled PMs was evaluated in mouse serum for 21 h, showing radiochemical stability above 85%. After in vivo evaluation of the 211At- labeled PMs, 4-5 % ID/g of the 211At could still be detected in the blood, showing a promising in vivo stability of the PMs. Further, 211At-labeled PMs accumulated in the spleen (20-30 %ID/g) and the liver (2.5- 5.5 %ID/g), along with some detection of 211At in the thyroid (3.5-9 %ID/g). This led to the hypothesis that deastatination takes place in the liver, whereas good stability of the 211At core-radiolabel was observed in the blood.
Keywords: Astatine-211, polymeric micelles, PEG-PLGA, alpha-therapy, iodine-125
1. Introduction
In recent years, the interest in α-particle emitting radionuclides for cancer therapy has increased dramatically [1,2]. This is due to their short travel distance in tissue of 50-100 µm, causing highly localized damage to targeted cancer cells, reducing off-target irradiation. The comparatively high linear energy transfer (LET) of α-particles leads to double-stranded breaks (DSBs) and clustered damage to the DNA [3-5]. This means that α-radiation can be effective in patients where treatment with β or γ radiation has failed [6,7]. Of the around 400 observed α-emitting radionuclides, only a small minority of them have suitable properties for clinical use. Currently, the most popular are thorium-227 (227Th), actinium-225 (225Ac), radium-223 (223Ra), bismuth-213 (213Bi), lead-212 (212Pb) and astatine-211 (211At) [8].
Astatine-211 (211At) arguably has the most promising properties, due to its relatively short half-life of 7.2 h, cheap and practical cyclotron-based production, and its suitable two-way decay chain. 58% of 211At decays to polonium-211 (211Po), which has a half-life of 0.52 s and decays further to stable lead-207 (207Pb) via emission of one α-particle. In the second pathway, 211At decays to bismuth-207, through the emission of one α-particle, which in turn also decays to stable 207Pb [8]. The combined result of the two decay pathways is 100% alpha-particle emission per decay of 211At, all in close proximity to the targeted cell.
The major challenge with 211At is the chemistry of this element. It shares some chemical similarity with the other halogens, especially iodine. However, the alkyl-211At bond and the aryl-211At bond are significantly weaker compared to the other halogens (Table 1). This is mainly related to the difference in size and the additional 5f electrons of 211At [9]. Nevertheless, Zalutsky and co-workers employed electrophilic destannylation on succinimidyl benzoate to make succinimidyl‐ astato‐benzoate (SAB) and conjugated this activated prosthetic group to antibodies [10,11]. Parallel strategies were since utilized by various other groups [12,13]. However, deastatination is still routinely observed [14]. This challenge has been addressed for instance via small molecules [15,16] and through nanoparticles (NPs). Utilizing its metallic character, stable 211At surface labeled gold nanoparticles (AuNPs) were reported by Bilewicz and co-workers [17,18], which later also showed excellent in vivo stability [19]. To the same end, chelated rhodium coordination centers [20] or silver nanoparticles [21,22] have been used.
Estimated bond strength between halogen and aryl or alkyl (reproduced from Coenen 1983)[50]
| X= | Fluorine [kJ/mol] | Chlorine [kJ/mol] | Bromine [kJ/mol] | Iodine [kJ/mol] | Astatine [kJ/mol] |
|---|---|---|---|---|---|
| CH3-X | 444 | 339 | 285 | 222 | 163 |
| C6H5-X | 523 | 398 | 335 | 268 | 197 |
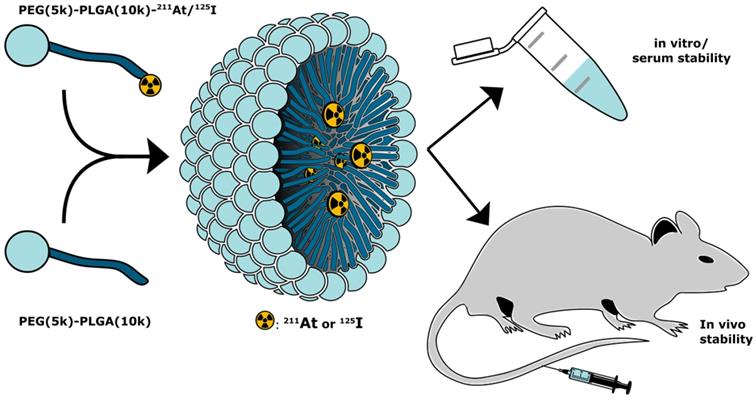
In the present report, we expand the NPs approach with a novel strategy for covalent radio-astatination and radio-iodination of the hydrophobic core of polymeric micelles (PMs, Figure 1). We used block copolymer PMs consisting of hydrophilic polyethylene glycol (PEG) and hydrophobic poly(lactic-co-glycolic acid) (PLGA), both of which are approved by the food and drug administration (FDA) for human use. By labeling the hydrophobic PLGA core of the PMs, we envisioned the protection of the weak 211At-aryl bond from metabolism. The PEG corona would ensure water solubility and biocompatibility [23-25], and would further allow for potential decoration with targeting ligands [26,27]. In this study, we report the synthetic and radiopharmaceutical strategy for preparing these PMs. Furthermore, we report high stability in mouse serum and in phosphate-buffered saline (PBS). Finally, the stability and biodistribution of 211At- core labeled PMs (211At-PMs) was investigated in vivo and compared to the biodistribution of free 211At.
Conceptual overview. Radiolabeled PEG-PLGA polymer was combined with non-modified PEG-PLGA polymer in a 1:10 ratio, to achieve polymeric micelles core-labeled with 125I or 211At (125I-PMs or 211At-PMs). The stability and biodistribution of these micelles were evaluated in vitro and in vivo in mice.
2. Results and Discussion
2.1 Indirect synthon-based radio-halogenation of PEG(5k)-PLGA(10k)
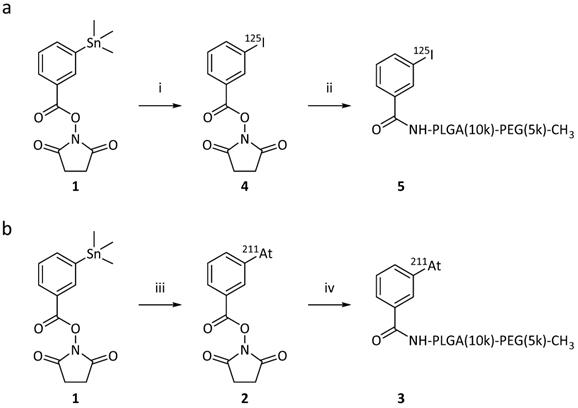
We employed two methods for radio-halogenation of the PEG(5k)-PLGA(10k)-NH2 copolymer; indirect and direct. Radiolabeled polymer would then be mixed with unlabeled copolymers to furnish radiolabeled PMs (Figure 1). The “indirect” method was a synthon-based approach. Here, the synthesis of polymers labeled with 125I (Scheme 1a) or 211At (Scheme 1b) was initiated from the well-established succinimidyl stannyl benzoate precursor (1) (Supplementary Information, S1). To activate the radioiodine, two equivalents of aqueous solution of the oxidative agent chloramine-T (CAT) were used, resulting in a radiochemical conversion (RCC) to compound [125I]4 of 95% determined by radioactive thin-layer chromatography (radio-TLC). Non-radioactive sodium iodide was subsequently added to quantitatively convert the remaining succinimidyl stannyl benzoate to the iodide derivative [127I]4. The same procedure was also attempted for radiolabeling with 211At. This failed however, and no conversion to SAB (2) was detected. Therefore, the well-established oxidants, N-iodosuccinimide (NIS) [28] and N-chlorosuccinimide (NCS) were evaluated instead of CAT (Scheme 1b). Both of these led to SAB (2) in quantitative (>95%) RCC (further details in Supplementary Information, S2). To convert the remaining succinimidyl stannyl benzoate (1) to the iodide [127I]4, NIS was added. The resulting product was analyzed via radio-TLC and compared to the retention factor (Rf) of the iodinated compound (4), which gave the same Rf.
Reaction schemes for the synthesis of the radiolabeled PEG-PLGA block co-polymers, via a synthon based approach (“indirect labeling”). The radiolabeling of the synthon was achieved by electrophilic destannylation. This was followed by the conjugation via nucleophilic substitution of the activated ester with the NH2-PLGA(10k)-PEG(5k). (a) Procedure used for iodination. (i) 1) [125I]NaI, CAT, AcOH, MeOH, rt, 15 min 2) NaI; (ii) 1) NH2PLGA(10k)-PEG(5k)-CH3, DIPEA, DMF, 50 °C, 2-3 h, 2) Ac2O, 50 °C, 1 h. (b) Procedure used for astatination. (iii) 1) [211At]At, NIS, AcOH, MeOH, rt, 15 min 2) NIS; (iv) 1) NH2PLGA(10k)-PEG(5k)-CH3, DIPEA, DMF, 50 °C, 2-3 h, 2) Ac2O, 50 °C, 1 h.
For both 125I and 211At, the crude reaction mixtures of 2 and 4 were dried without any further purification and subsequently dissolved in dimethylformamide (DMF). N,N-diisopropylethylamine (DIPEA) as well as PEG(5k)-PLGA(10k)-NH2 were added to the reaction mixtures. The conjugation reaction was continued until no radiolabeled synthon (2 or 4) could be detected via radio-TLC. No formation of iodo- or astatobenzoic acid, resulting from NHS ester hydrolysis, was observed by radio-TLC. Acetic anhydride was added in excess to convert unreacted amines to the corresponding acetamides in order to prevent highly cationic final PMs. By slow addition of water over 2 min, the formation of radio-halogenated PMs could be observed, through the appearance of opalescence in the mixture. The radio-halogenated PMs were purified with a centrifuge filter by repeated steps of centrifugation and redispersion in freshly added water. The final radiolabeled PEG(5k)-PLGA(10k) (3 or 5) were then dried completely under nitrogen flow for 10-30 min. In the case of the PEG(5k)-PLGA(10k)-125I (5) an overall radiochemical yield (RCY) of 36% was achieved, whereas the overall RCY for the PEG(5k)-PLGA(10k)-211At (3) was 32% (decay-corrected).
2.2 Direct radio-halogenation of PEG(5k)-PLGA(10k)
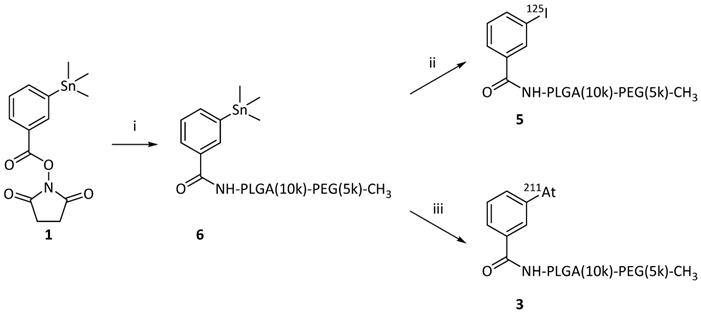
As the second method for synthesizing the radio-halogenated PEG(5k)-PLGA(10k) (3 or 5), a “direct” approach was chosen (Scheme 2). This procedure had the advantage that only the final step involved handling radioactive materials. In this case, the succinimidyl stannyl benzoate (1) was first reacted with the PEG(5k)-PLGA(10k)-NH2 polymer. Completion of the reaction was monitored via consumption of the starting material (1) by TLC. The remaining amines were then capped by acetic anhydride addition with complete capping validated via a Kaiser test, showing the desired lack of development of blue color in all cases. After purification on a centrifuge filter as described above, the stannylated PEG(5k)-PLGA(10k) (6) was lyophilized to provide an amorphous solid. For the final radio-halogenation step, similar radiochemical procedures as described above were used. For the radio-iodination, chloramine-T was used as oxidative agent and quantitative radiolabeling was achieved within 15 min. For the astatination, NCS was used to activate the 211At. The so-achieved radio-halogenated PEG(5k)-PLGA(10k) (3 or 5) were again purified with a centrifuge filter and dried under nitrogen, as described above. This resulted in RCYs of 72% for 211At and 59% for 125I.
Reaction schemes for the synthesis of the radiolabeled PEG-PLGA block co-polymers, via the “direct” on-polymer destannylation approach. Procedure used for iodination and for astatination of the PMs. (i) 1) NH2-PLGA(10k)-PEG(5k)-CH3, DIPEA, DMF, rt, overnight, 2) Ac2O, rt, 2h;(ii) 1) [125I]NaI, CAT, AcOH, DMF, rt, 15 min 2) NaI;.(iii) [211At]At, NCS, AcOH, DMF, rt, 15 min.
2.3 Preparation of the radio-halogenated PMs
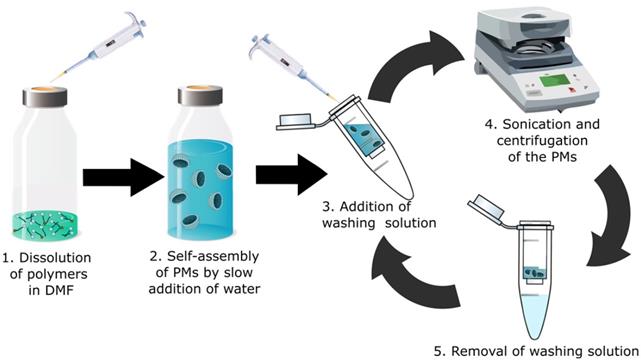
For the preparation of the radio-halogenated PMs, a rapid and practical method was needed, due to the short 7.2 h half-life of 211At. The most common conventional method for PM preparation is extended dialysis after self-assembly [29-31]. This procedure typically takes 24-48 h and is not suitable for a radionuclide like 211At. For that reason, a new procedure to rapidly form the micelles was developed (Figure 2). The radio-halogenated PEG(5k)-PLGA(10k) (3 or 5) were mixed with non-radiolabeled PEG(5k)-PLGA(10k) to obtain a ratio of radio-halogenated PEG(5k)-PLGA(10k) (3 or 5) corresponding to a maximum of 5-10%. The resulting mixture was dissolved in DMF and water was added slowly to achieve a final PEG(5k)-PLGA(10k) concentration of 3 mg/mL and a DMF concentration in the final PM dispersion below 3%. Through this procedure, PEG(5k)-PLGA(10k) self-assembles to spherical PMs due to the hydrophophicity of PLGA and the hydrophilicity of PEG [32]. Furthermore, the hydrophilic shell makes the micelles dispersible in aqueous media, and gives protection from the mononuclear phagocyte system (MPS), enabling extended circulation in the bloodstream [33].
Schematic overview of the optimized rapid preparation of radio-halogenated PMs; 1. A 1:10 mixture of radiolabeled PEG-PLGA with non-labeled PEG-PLGA is dissolved in DMF. (2) Addition of water (50:1 ratio to DMF) to form PMs. (3) Addition of PBS or water as washing solution. (4) Sonication for 20 min at RT and subsequent centrifugation in a centrifuge filter. (5) Removal and discarding of washing solution (filtrand) containing potential impurities and traces of DMF. The washing cycle is repeated 3-5 times.
After PM formation, additional purification steps were performed to remove residual DMF from the dispersion. Purification in a centrifuge filter (30 kDa) and sonication at room temperature were repeated four times to achieve radio-halogenated PMs with an intensity weighted diameter of 139.8 ± 8.5 nm (Table 2). No significant difference in size between 211At-PMs and 125I-PMs was found. In addition, cryo-transmission electron microscopy (cryo-TEM) analyses of the 211At-PMs after the 211At was decayed were carried out (Supplementary Information S3 A). To assess if any radioactive species could still be separated from the radio-halogenated PMs, a PD-10 size-exclusion purification step was carried out (Supplementary Information, S4). In all cases, 83 ± 4% of the radioactivity was found in the large molecular fraction containing the radio-halogenated PMs, indicating successful formation of 211At-PMs and 125I-PMs. The formulation and purification was achieved within 2 h, which is less than a third of the half-life of 211At. This reduced processing time is vital for the application in combination with 211At and allows for potential further modification. In addition, this fast approach could be used to further alter the PM composition and thereby for example add a targeting vector to the surface of the PMs.
Properties of final 125I or 211At core-radiolabeled PMs, as measured by DLS (n = 3)
| Mean Diam. by Number [nm] | Mean Diam. by Volume [nm] | Mean Diam. by Intensity [nm] | PDI | Zeta potential [mV] |
|---|---|---|---|---|
| 67 ± 15 | 83 ± 14 | 140 ± 9 | 0.16 ± 0.02 | -10.9 ± 0.9 |
2.4 In vitro stability of the 211At-PMs
The stability of both directly and indirectly labeled 125I-PMs and 211At-PMs was assessed in PBS at 37 °C over 4 h with the same procedure (Supplementary Information, S5). Since both exhibited excellent radiochemical stability at >95% based on the amount of 125I or 211At detected in the washing fraction (filtrate), the focus was shifted towards the therapeutically more relevant 211At-PMs. For these PMs, an extended stability evaluation in PBS was performed for 21 h. After incubation in PBS, three washing steps in a centrifuge filter (30 kDa) were carried out to separate smaller radioactive species resulting from degradation of the 211At-PMs. Aliquots of both washing fractions (filtrates) and the 211At-PMs were measured on a gamma counter and compared to the total added radioactivity (Figure 3a). 80-99 % of the initial radioactivity was accounted for with losses attributed to 211At-PMs adsorbing to the filter. This was supported by non-radioactive control experiments, in which, PMs were formed and washed five times in the centrifuge filters. The filtrand was then dried and the residue weighed, giving 81.6 ± 15.6% of the total polymer mass. In the case of stability in PBS of the indirectly labeled 211At-PMs, 95% of the radioactivity was found in the PM containing fraction. A similar result was observed for the directly labeled 211At-PMs, where 94.2 ± 6.5 % of the 211At was found in the PM containing fraction. For the control group, the opposite result was seen. Only 1.0 ± 0.1% of free 211At was measured in the filter. This supports excellent stability in PBS of both directly and indirectly labeled 211At-PMs.
In vitro stability of 211At-PMs in (a) PBS (n = 3) or (b) mouse serum (n = 3). After an incubation time of 21 h in PBS or mouse serum, the 211At-PMs were separated from the free 211At with a centrifuge filter to achieve a fraction with the 211At-PMs (filtrand) and the washing fraction (filtrate) containing free 211At. The results were compared to a control group, where free 211At was incubated with (a) PBS (n = 3) or (b) mouse serum (n = 3). The results were expressed in % of the total activity measured before separation.
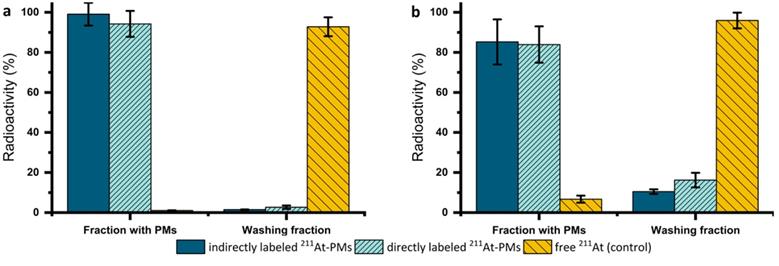
To evaluate stability in biomimetic conditions, 211At-PMs were also incubated in mouse serum mixed with PBS (2:1) for 21 h and the same analytical procedure described above was used (Figure 3b). To ensure that the procedure successfully separated released free 211At from biological material, a control experiment was performed in which free 211At was incubated and treated in the same way. In the serum stability test for directly labeled 211At-PMs, 16.2 ± 3.6 % (n = 3) of the activity was found in the filtrate. This is significantly (p = 0.15 analyzed by t-test) more than for indirectly labeled 211At, where only 10.5 ± 1.1% (n = 3) was found in the filtrate, indicating a more compromised stability of the indirectly labeled 211At-PMs. The direct labeling procedure has one main disadvantage in comparison to the indirect labeling method, which is the challenge of monitoring the chemical form of the radionuclide during the radiolabeling procedure and ensuring conversion to the desired product. The direct labeling takes place on the polymer, which cannot as easily be resolved chromatographically on standard radioanalytical equipment, such as radio-TLC and radio-HPLC. The radioactivity can most practically be followed by size exclusion chromatography and by tracking of the radiolabeled PMs themselves. This cannot with certainty exclude that small molecular 211At species could be non-covalently retained inside the micellar core. Such 211At species could potentially leak slowly from the 211At-PMs, giving rise to an observed instability. In contrast to that, for the indirect labeling, each radio synthetic step could be accurately monitored by radio-TLC (Supplementary Figure S1). Thus, it could be assured that all the added 211At is covalently bound to the precursor (2) in the first step and then attached to the polymer (3). Concluding from that, it is likely that the differences between the groups of around 6% of the activity can be ascribed to non-covalently bound 211At in the case of the directly labeled 211At-PMs. Accordingly, with a short-lived radionuclide like 211At, direct labeling is the preferred method, whereas for long-lived nuclides such as 125I, one should primarily consider the indirect method.
2.4 In vivo stability evaluation of the 211At-PMs in mice
An in vivo stability study was conducted with both directly and indirectly radiolabeled 211At-PMs in healthy Balb/c mice. In all experiments, an activity of 0.4-0.6 MBq was injected trough the tail vein. Mice were sacrificed at 21 h post-injection and excised organs were measured for radioactivity content on a gamma counter (Figure 4). All data were compared to the biodistribution of free 211At, for which data was already available from a recent report from our group [19]. In this way, indirect evidence of the stability of the aryl-211At can be obtained, including where the deastatination takes place. The 21 h time-point was chosen, since widespread uptake in liver and spleen would already be manifest at this point, with potential 211At release as a result, and at the same time with appreciable remaining blood circulation [34-36].
Ex vivo biodistribution at 21 h post-injection of 211At-PMs in healthy Balb/c mice (n = 3). The results from the PMs are compared to data for free 211At administered in the same mouse model generated in a recent report by our group [19]. The data were analyzed and compared to each other by a one-way ANOVA followed by a Tukey's multiple comparison test with the following significance thresholds (*) p > 0.1, (**) p > 0.05, (***) p < 0.01.
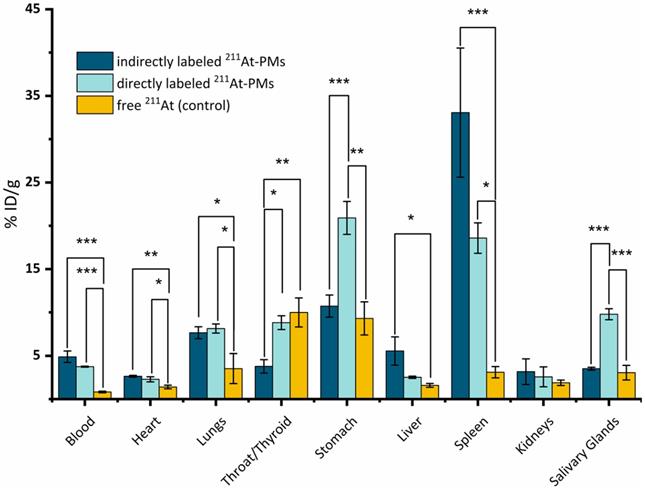
Overall, highly significantly different organ uptakes were observed between the free 211At data and the two 211At-PMs groups. In some cases, a significant difference between the two 211At-PM groups could also be observed. For the free 211At control group at 21 h, 10.0 ± 1.7% ID/g was previously reported to accumulate in the thyroid and 9.3 ± 1.7% ID/g in the stomach. Further, 3.5 ± 1.7% ID/g was found in the lungs. These observations correlate with other previous reports [37,38].
In the blood, only small amounts of free 211At were present at 21 h in the control data (0.8 ± 0.1% ID/g). In the case of both indirectly and directly labeled 211At-PMs, a different picture was observed. In both cases, around 4-5% (indirect: 4.9 ± 0.7% ID/g and direct: 3.8 ± 0.1% ID/g) of the 211At was still present in the blood stream at 21 h. This demonstrated that the 211At was long circulating when associated with the PMs, supporting that the two core-radiolabeling strategies are stable in the bloodstream and protect the 211At-radiolabel from deastatination during circulation. As a primary site for nanoparticle accumulation due to uptake by macrophages, PMs are expected to accumulate in the liver.[39]. However, we only observed 5.5 ± 1.6% ID/g and 2.5 ± 0.2% ID/g liver uptake for directly and indirectly labeled 211At-PMs, respectively. Conversely, uptake in the spleen was high, at 18.6 ± 1.8% ID/g and 33.1 ± 7.5% ID/g for directly and indirectly labeled 211At-PMs, respectively, compared to just 3.1 ± 0.7% ID/g for free 211At. It is known that nanoparticles in the 50-200 nm size range accumulate in the spleen, making the observed splenic accumulation a hallmark of nanoparticle behavior. It can therefore be hypothesized that the lack of observed hepatic accumulation is related to degradation after uptake by liver macrophages. Thus, the PMs appear to be destabilized after macrophage uptake, with the 211At-aryl bond to an extent broken, potentially via oxidation as has been reported to be a source of deastatination [14]. As 211At is non-residualizing, this would lead to release into the bloodstream and accumulation of free 211At in organs such as thyroid and stomach, as was observed for the free 211At control. This can therefore also explain the high accumulation for both PM groups in the stomach (indirect: 10.7 ± 1.3% ID/g and direct: 20.9 ± 1.9% ID/g) as well as in the thyroid (indirect: 3.8 ± 0.8% ID/g and direct: 8.8 ± 0.8% ID/g). Comparing with literature reports on antibodies and antibody fragments labeled with SAB, lower stomach accumulation values of 5-8 %ID/g at 22-24 h was observed in these reports [40,41]. We suggest that this difference could be a result of less intrinsic liver accumulation of antibodies and fragments than for nanoparticles, resulting in less deastatination. In our previous study on 211At-labeled AuNPs we observed higher liver accumulation at around 40-55 %ID/g suggesting higher stabillity towards deastatination in the liver for the AuNP surface label than for these PMs [19]. However, in a setting of locoregional administration, this difference is likely to be less pronounced, with PMs having the advantage of being biodegradable.
For both stomach and thyroid, significantly more 211At was measured for the directly labeled PMs. This could be due to the difference in the labeling method. As already indicated by the mouse serum stability studies above, the direct labeling method could potentially contain non-covalently trapped 211At in the core of the PMs, which could be released under the right conditions. This could lead to a higher concentration of free 211At when compared to the indirectly labeled PMs, and therefore to a higher accumulation in the thyroid and stomach.
Although deastatination seemed to occur in the liver, an appreciable stability was observed in the blood as well as in the spleen. Concluding from that, the herein reported 211At-PMs, while potentially not ideal for systemic administration, could be valuable for locally administered tumor treatment. This approach has been utilized in several reports and has already shown effective results when labeling antibodies with 211At [42,43]. An additional possible use for NPs labeled with 211At could be brachytherapy as shown in a recent report, where tumor growth was inhibited for 38 days [44]. When administering the 211At-PMs directly at the tumor site, accumulation in the liver can be avoided and, therefore, the degradation of the micelles and consequent deastatination may be limited. In addition to 211At, a combination with diagnostic radionuclides like 123I, 124I or even 18F could be imaginable, due to the fast preparation method of the final PMs described in this manuscript. This theranostic approach, where the therapeutic radionuclide and the diagnostic radionuclide are combined in the same particle, holds high potential in nuclear medicine. To attain this, the stability of the PMs as well as their size should be optimized. One way to do that is by crosslinking the micelles, which increases their overall stability [45].
3. Conclusions
We report a strategy for covalent core-radiolabeling of PMs with 211At and 125I. The labeling was achieved via two different strategies, direct and indirect. In “indirect” labeling, a succinimidyl stannyl benzoate precursor (1) was labeled with 211At or 125I and conjugated to the polymer PEG(5k)-PLGA(10k)-NH2, followed by PM formation. For the “direct” labeling, the succinimidyl stannyl benzoate (1) was first conjugated to the polymer, followed by labeling with 125I or 211At. Due to the short half-life of 211At (7.2 h), a rapid method for forming PMs was developed, taking approximately 2 h. 211At-PMs were tested in mouse serum and exhibited a stability of >85% after 21 h. Ex vivo biodistribution in mice showed that after 21 h, 4-5% ID/g still circulated in the blood, suggesting appreciable stability. However, only small amounts of 211At were detected in the liver, indicating deastatination after uptake by liver macrophages. Uptake of 211At in thyroid and stomach also suggested partial deastatination. Outside the liver, the 211At-PMs appeared stable, and may be utilized for local therapy in a post-surgery or disseminated disease setting.
4. Materials and Methods
Materials. Milli-Q (MQ) water (18.2 MΩ × cm) was used for all preparation steps. All chemicals were purchased from Sigma Aldrich beside the PEG(5k)-PLGA(10k) and the PEG(5k)-PLGA(10k)-NH2 which were acquired form Nanosoft Polymers. The size was measured four-five times on the same sample and reported as mean square displacement (MSD) calculated averages. DLS measurements were carried out in the reaction mixture medium without dilution (sodium citrate and Milli-Q water) or PBS buffer at 25 °C. Radio-TLC plates were analysed using a Ray test MiniGita apparatus equipped with a Beta detector GMC, or a Perkin Elmer Cyclone® Plus Storage Phosphor System. Radioactivity was quantified with a Princeton Gammatech LGC 5 germanium detector Gamma spectrometer. Activity was measured on a dose calibrator (VEENSTRA instruments or CRC-55tR). The [125I]NaI was purchased from Perkin Elmer.
Production of 211At. 209Bi(α,2n)211At reaction was used to produce 211At, starting from 209Bi deposited on an aluminium backing. The production was performed at 29 MeV with α-particles using a Scanditronix MC32 cyclotron at the PET & Cyclotron Unit at Copenhagen University Hospital, Denmark. The irradiated target was purified at the the Department of Nuclear Medicine, Sahlgrenska University Hospital, Sweden by dry distillation as previously reported. [46]
2,5-Dioxopyrrolidin-1-yl 3-iodobenzoate. Dissolution of 3-Iodobenzoic acid (4.86 g, 19.62 mmol, 1.05 equiv) in dry THF (75 mL) was followed by addition of DCC (3.85 g, 18.68 mmol, 1.00 equiv) and N-hydroxysuccinimide (2.15 g, 18.68 mmol, 1.00 equiv) to the suspension. Stirring at room temperature for 20 h ensued and the reaction mixture was subsequently filtered. The clear filtrate was concentrated under reduced pressure to obtain a solid, which was dissolved in DCM (50 mL) and washed with aq. Na2CO3 (2 M, 2 × 20 mL). Drying of the organic phase over MgSO4 and purification of the crude product by CombiFlash (EtOAc in heptane, 0 to 55% v/v), yielded a white solid (4.21 g, 12.20 mmol, 65%). Rf = 0.45 (1:1 EtOAc/heptane); 1H NMR (600 MHz, CDCl3) δ 8.47 (t, J = 1.8 Hz, 1H), 8.10 (dd, J = 7.9, 1.4 Hz, 1H), 8.01 (dd, J = 7.9, 1.6 Hz, 1H), 7.26 (t, J = 7.9 Hz, 1H), 2.91 (s, 4H); 13C NMR (151 MHz, CDCl3) δ 169.1, 160.7, 143.9, 139.3, 130.6, 129.8, 127.2, 94.1, 25.8 and is in agreement with previously published data [47]
2,5-Dioxopyrrolidin-1-yl 3-(trimethylstannyl)benzoate (1). An evacuated and argon purged microwave vessel was prepared and 1,1,1,2,2,2-hexamethyldistannane (300 μL, 1.45 mmol, 2.00 equiv) in dry, degassed THF (5 mL) was added. 1,3,5,7-tetramethyl-2,4,8-trioxa-(2,4-dimethoxyphenyl)-6-phosphaadamantane (meCgPPh) (21.2 mg, 72.5 μmol, 0.10 equiv) and Pd(OAc)2 (8.1 mg, 36.2 μmol, 0.05 equiv) were further included and the vessel was sealed. Stirring at room temperature was implemented for 5 min. Then, 2,5-dioxopyrrolidin-1-yl 3-iodobenzoate (250 mg, 0.72 mmol, 1.00 equiv) in dry, degassed THF (5 mL) was added under argon flow. The microwave vessel was heated in the microwave for 30 min at 70 °C. After cooling and concentrating under reduced pressure, the reaction mixture was purified by CombiFlash (EtOAc in heptane, 0 to 40% v/v), yielding a viscous oil, which slowly solidified (721 mg, 0.71 mmol, 98%). Rf = 0.31 (2:1 EtOAc/heptane); 1H NMR (400 MHz, CDCl3) δ 8.30 - 8.16 (m, 1H), 8.07 (dt, J = 8.0, 1.6 Hz, 1H), 7.79 (dt, J = 7.3, 1.1 Hz, 1H), 7.47 (td, J = 7.9, 3.7 Hz, 1H), 2.91 (s, 4H), 0.34 (s, 8H); 13C NMR (101 MHz, CDCl3) δ 169.4, 162.5, 143.9, 142.4, 137.8, 130.4, 128.3, 124.7, 25.9, -9.3 and is in agreement with previously published data [48,49]
2,5-dioxopyrrolidin-1-yl 3-(astato-211At)benzoate (2). Stock solutions of all compounds were freshly prepared. N-iodosuccinimide (4.55 mg, 0.02 mmol) was dissolved in dry methanol (450 µL) with 1% AcOH and diluted 1:100 with methanol containingh 1% AcOH), to get a final concentration of 0.0001 mg/µL. 2,5-Dioxopyrrolidin-1-yl 3-(trimethylstannyl)benzoate (1) (1.84 mg, 0.0048 mmol) was dissolved in methanol (1.84 mL) with 1% AcOH and diluted with methanol containing 1% AcOH to achieve a concentration of 0.001 mg/µL. 211At (20 MBq) in chloroform was dried under constant nitrogen flow and dissolved in methanol containing 1% AcOH (30 µL). To this solution, the NIS-stock solution (13 µL, 0.0013 mg, 0.0057 µmol, 0.1 eq) was added under vigorous stirring. Precursor 1 stock solution (22 µL, 0.022 mg, 0.057 µmol, 1 eq.) was added to the reaction mixture and analysed via radio-TLC (1:1 EtOAc:Hexan) after 15 min, showing a radiochemical conversion (RCC) of 95%. After a further 5 min, 13 µL of a 10 times more concentrated NIS-stock solution (0.013 mg, 0.057 µmol, 1 eq) was added to the reaction mixture and stirred for 30 min at room temperature. The mixture was dried at 50 °C under constant nitrogen flow and used for the next step without further purification.
3-(astato-211At)-benzamide- N-(PEG(5k)-PLGA(10k)) (3). DIPEA (5.8 µL, 4.3 mg, 0.03 mmol) was dissolved in dry DMF (500 µL) and diluted with DMF 1:10 for a concentration of 0.86 mg/mL. PEG(5k)-PLGA(10k)-NH2 (2 mg, 0.14 µmol) was dissolved in the DIPEA-stock solution (100 µL). 50 µL of this solution, containing PEG(5k)-PLGA(10k)-NH2 (1 mg, 0.07 µmol, 1 eq) and DIPEA (0.043 mg, 0.33 µmol), was added to the vial containing the radiolabeled precursor (2). The mixture was heated to 50 °C and the progress of the reaction was followed by radio-TLC. After 2-3 h, starting material (2) was no longer detected. Acetic anhydride (1 µL) was added and stirred at 50 °C for a further hour. After this mixture had cooled to room temperature, water (1.0 mL) was added over 2 min, where the appearance of opalescence indicated the formation on PMs. This reaction mixture was transferred to an Amicon® centrifuge filter (30 kDa) and centrifuged at 4.4 krpm for 10 min. Water (2.0 mL) was then added to the filter containing the polymer product 3, mixed and the centrifugation was repeated. This procedure was then repeated a further 4-5 times. The resulting polymer solution in the filter was transferred to an HPLC vial and dried under constant N2-flow at 50 °C.
2,5-dioxopyrrolidin-1-yl 3-(iodo-125I)benzoate (4). A stock solution with a concentration of 1 mg/mL of 2,5-dioxopyrrolidin-1-yl 3-(trimethylstannyl)benzoate (1) in methanol was prepared. A further stock solution of 5 mg/mL of chloramine-T in water was prepared. To methanol (60 µL), AcOH (8 µL) was added. To the solution of 1 in MeOH was added [125I]NaI (25 MBq) in aq. NaOH (0.1 M, 3 µL) and the aq. chloramine-T solution (9 µL, 0.045 mg, 0.160 µmol, 2 eq.). Under vigorous stirring, 3 µL of the 2,5-dioxopyrrolidin-1-yl 3-(trimethylstannyl)benzoate(1)-solution (0.03 mg, 0.08 µmol, 1 eq) was added and the mixture was analyzed by radio-TLC after 10 min. The remaining stannyl moiety on 1 was quenched by adding aq. NaI (3 µL, 0.012 mg, 0,08 µmol) and stirred for a further 15 min. The mixture was dried and directly used for conjugating to the PEG(5k)-PLGA(10k)-NH2.
3-(iodo-125I)-benzamide-N-(PEG(5k)-PLGA(10k)) (5). The same procedure as for compound 3 was used.
3-(trimethylstannyl)benzamide-N-PEG(5k)-PLGA(10k) (6). DIPEA (5.8 µL , 4.3 mg, 0.03 mmol) was dissolved in DMF (5 mL) to achieve a concentration of 0.86 mg/mL. 200 µL of this solution was used to dissolve PEG(5k)-PLGA(10k)-NH2 (10 mg, 0.67 µmol, 1 eq). Stannyl precursor 1 (0.21 mg, 0.54 µmol, 0.8 eq) was dissolved in DMF (50 µL) and mixed with the solution of PEG(5k)-PLGA(10k)-NH2. The resulting reaction mixture was stirred overnight at room temperature. A TLC analysis was performed to confirm complete consumption of 1. Acetic anhydride (1 µL) was added and the mixture was stirred for a further 2 hours. The capping of the remaining amines was confirmed by a Kaiser test. Water (1.0 mL) was added to the reaction mixture, which was then purified on a centrifuge filter by centrifugation (4.4 krpm, 10 min) and redispersion in water (3 mL), which was repeated five times. The final solution was lyophilized to provide the final polymer 6 in a range of 7-9 mg.
3-(astato-211At)-benzamide-N-PEG(5k)-PLGA(10k) (3). A stock solution of N-chlorosuccinimide (NCS) of 5 mg/mL in methanol was prepared. 3-(trimethylstannyl)benzamide-N-PEG(5k)-PLGA(10k) (6) (0.3 mg, 0.02 µmol) was dissolved in DMF (30 µL) and transferred to a vial containing 12 MBq dry 211At. Further DMF (20 µL) and AcOH (0.5 µL) was added to the reaction mixture. After 1 min, 10 µL of the NCS-stock solution was added and was stirred for 30 min. To the mixture, water (1 mL) was added slowly and transferred to an Amicon® centrifuge filter. The vial was washed with water (2 mL) and added to the centrifuge filter. The solution was centrifuged at 4.4 krpm for 10 min and water (3 mL) were added. The washing procedure was repeated twice more. The so achieved solution was transferred to a vial and dried at 50 °C under constant N2-flow.
3-(iodo-125I)-benzamide-N-(PEG(5k)-PLGA(10k)) (5). Same procedure as described for 3-(astato-211At)-benzamide-N-PEG(5k)-PLGA(10k) (3) was used as described above.
Preparation of PMs. The radiolabeled polymer (3, 5, 7 or 8) was mixed with PEG(5k)-PLGA(10k) (3-9 mg, 0.2-0.6 µmol) to achieve a concentration of 5-10 w/w% radiolabeled polymer (3, 5, 7 or 8). This mixture was dissolved in DMF (30-60 µL) over 30 min. To this mixture, water (1-3 mL) was added slowly under shaking to aim for a final concentration of 3 mg/mL. The mixture was then transferred to a centrifuge filter and centrifuged for 10 min at 4.4 krpm. PBS (3 mL) was added to the solution in the filter and mixed with the pipette 5 times. The filter was closed with para film and put into the ultrasonic bath for 20 min at room temperature. Afterwards, they were centrifuged again and the entire washing procedure was repeated twice more in order to achieve the final micelles in PBS.
Characterization by DLS. The size and the zeta potential of the core-radiolabeled PMs were measured via DLS. For the 125I-PMs the size and the zeta potential in PBS was measured directly after the PMs were formed and purified. The size and zeta-potential of the 211At-PMs in PBS was measured after the 211At was decayed.
PBS Stability study. 100 kBq of the core labeled PMs (100 µL) were diluted to 1.0 mL to achieve a concentration of 100 kBq/mL. These PMS were shaken for 21 h and afterwards filtered on the centrifuge filter at 4.4 krpm for 10 min. The PMs were washed three more times with water (1 mL) by Amicon centrifuge filter (30 kDa) and an aliquot of the fraction in the filter and in the filtrate was measured on the γ-counter. The results were expressed as radioactivity compared to total radioactivity in percent.
Serum stability. To 100 kBq of the direct and indirect labeled PMs (100 µL), mouse serum (200 µL) was added. This was shaken for 21 h. Afterwards, the mixture was transferred to the centrifuge filter and diluted with 1 mL PBS. This mixture was centrifuged at 4.4 krpm for 10 min. To remove all of the free 211At from the PMs, to the mixture PBS (1 mL) was added and centrifuged at 4.4 krpm for 10 min with Amicon centrifuge filter (30 kDa). This step was repeated 3. An aliquot from the fraction in the filter and from the washing fraction was measured on the gamma counter.
In vivo stability study. The 211At labeled PMs were concentrated using an Amicon® centrifugal filter device (10 min, 4.4 krpm) and then washed three times with PBS. Resuspension in PBS to attain a radioactivity concentration of 0.5 MBq/100µL ensued. Evaluation of the biodistribution of all samples was carried out in healthy female Balb/C nu/nu mice (Janvier Labs, France), 4-6 weeks of age. This study was approved by the Gothenburg Ethical Committee for Animal Research (Ethical permit: 2138-19), and all animals were maintained as regulated by the Swedish Animal Welfare Agency. Housing of the mice was implemented in sterile conditions at 22 °C with access to food and water ad libitum. 0.5 MBq of 211At-PMs in 100 µL were injected intravenously through the tail vein six mice. 21 h after injection, the mice were sacrificed and blood was obtained by cardiac puncture and excision of tissues including thyroid/throat, salivary glands, heart, lungs, liver, stomach, kidneys, and spleen followed. Weighing of tissues and measuring of 211At on a NaI(Tl) γ-counter (Wizard 1480, Wallac, Finland) yielded results presented as percent of injected dose per gram of organ (%ID/g).
Abbreviations
AuNPs: gold nanoparticles; CAT: chloramine-T; DLS: dynamic Light Scattering; DIPEA: N,N-diisopropylethylamine; DMF: dimethylformamide; DSBs: double-stranded breaks; FDA: food and drug administration; LET: linear energy transfer; MPS: mononuclear phagocyte system; NCS: N-chlorosuccinimide; NIS: N-iodosuccinimide; NPs: nanoparticle; PBS: phosphate-buffered saline; PEG: polyethylene glycol; PLGA: poly(lactic-co-glycolic acid); PMs: polymeric micelles; RCC: radiochemical conversion; RCY: radiochemical yield; Rf: retention factor; SAB: succinimidyl astatobenzoate; TEM: transmission electron microscopy; TLC: thin-layer chromatography.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nelson BJB, Andersson JD, Wuest F. Targeted Alpha Therapy: Progress in Radionuclide Production, Radiochemistry, and Applications. Pharm. 2021;13:49
2. Tafreshi NK, Doligalski ML, Tichacek CJ, Pandya DN, Budzevich MM, El-Haddad G. et al. Development of Targeted Alpha Particle Therapy for Solid Tumors. Mol. 2019;24:4314
3. Sgouros G, Roeske JC, McDevitt MR, Palm S, Allen BJ, Fisher DR. et al. Radiobiology and Dosimetry of α-Particle Emitters for Targeted Radionuclide Therapy. J Nucl Med. 2010;51:311-328
4. Parker C, Lewington V, Shore N, Kratochwil C, Levy M, Lindén O. et al. Targeted Alpha Therapy, an Emerging Class of Cancer Agents: A Review. JAMA Oncol. 2018;4:1765-1772
5. Morgenstern A, Apostolidis C, Kratochwil C, Sathekge M, Krolicki L, Bruchertseifer F.An Overview of Targeted Alpha Therapy with 225 Actinium, 213 Bismuth. Curr Radiopharm. 2018;11:200-208.
6. Kratochwil C, Giesel FL, Bruchertseifer F, Mier W, Apostolidis C, Boll R. et al. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: a first-in-human experience. Eur J Nucl Med Mol Imaging. 2014;41:2106-2119
7. Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F. et al. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J Nucl Med. 2016;57:1941-1944
8. Lindegren S, Albertsson P, Bäck T, Jensen H, Palm S, Aneheim E. Realizing Clinical Trials with Astatine-211: The Chemistry Infrastructure. Cancer Biother Radiopharm. 2020;35:425-436
9. Meyer GJ. Astatine. J Label Compd Radiopharm. 2018;61:154-164
10. Zalutsky MR, Narula AS. Astatination of proteins using an N-succinimidyl tri-n-butylstannyl benzoate intermediate. Int J Radiat Appl Instrumentation Part A Appl Radiat Isot. 1988;39:227-232
11. Zalutsky MR, Reardon DA, Pozzi OR, Vaidyanathan G, Bigner DD. Targeted α-particle radiotherapy with 211At-labeled monoclonal antibodies. Nucl Med Biol. 2007;34:779-785
12. Andersson H, Cederkrantz E, Bäck T, Divgi C, Elgqvist J, Himmelman J. et al. Intraperitoneal α-Particle Radioimmunotherapy of Ovarian Cancer Patients: Pharmacokinetics and Dosimetry of 211At-MX35 F(ab′)2—A Phase I Study. J Nucl Med. 2009;50:1153-1160
13. Aneheim E, Gustafsson A, Albertsson P, Bäck T, Jensen H, Palm S, S. et al. Synthesis and Evaluation of Astatinated N-[2-(Maleimido)ethyl]-3-(trimethylstannyl)benzamide Immunoconjugates. Bioconjug Chem. 2016;27:688-697
14. Teze D, Sergentu DC, Kalichuk V, Barbet J, Deniaud D, Galland N. et al. Targeted radionuclide therapy with astatine-211: Oxidative dehalogenation of astatobenzoate conjugates. Sci Reports. 2017;7:1-9
15. Suzuki H, Kaizuka Y, Tatsuta M, Tanaka H, Washiya N, Shirakami Y. et al. Neopentyl Glycol as a Scaffold to Provide Radiohalogenated Theranostic Pairs of High In vivo Stability. J Med Chem. 2021;64:15846-15857
16. Denk C, Aneheim EHK, Lindegren S, Herth M, Wilkovitsch M, Mikula H. A Novel Multi-Component Reaction for At-211-Astatination: Providing new Tools for (pre) Targeted Alpha Therapy. In European Journal of Nuclear Medicine and Molecular Imaging. 2017;44:135-S136
17. Dziawer L, Koźmiński P, Mȩczyńska-Wielgosz S, Pruszyński M, Łyczko M, Wąs B. et al. Gold nanoparticle bioconjugates labelled with 211At for targeted alpha therapy. RSC Adv. 2017;7:41024-41032
18. Dziawer Ł, Majkowska-Pilip A, Gaweł D, Godlewska M, Pruszyński M, Jastrzębski J. et al. Trastuzumab-Modified Gold Nanoparticles Labeled with 211At as a Prospective Tool for Local Treatment of HER2-Positive Breast Cancer. Nanomater. 2019;9:632
19. Sporer E, Poulie CBM, Lindegren S, Aneheim E, Jensen H, Bäck T. et al. Surface Adsorption of the Alpha-Emitter Astatine-211 to Gold Nanoparticles Is Stable In vivo and Potentially Useful in Radionuclide Therapy. J Nanotheranostics. 2021;2:196-207
20. Lyczko M, Pruszynski M, Majkowska-Pilip A, Lyczko K, Was B, Meczynska-Wielgosz S. et al. 211At labeled substance P (5-11) as potential radiopharmaceutical for glioma treatment. Nucl Med Biol. 2017;53:1-8
21. Kučka J, Hrubý M, Koňák Č, Kozempel J, Lebeda O. Astatination of nanoparticles containing silver as possible carriers of 211At. Appl Radiat Isot. 2006;64:201-206
22. Cedrowska E, Lyczko M, Piotrowska A, Bilewicz A, Stolarz A, Trzcińska A. et al. Silver impregnated nanoparticles of titanium dioxide as carriers for 211At. Radiochim Acta. 2016;104:267-275
23. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chemie Int Ed. 2010;49:6288-6308
24. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polym. 2011;3:1377-1397
25. Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric Systems for Controlled Drug Release. Chem Rev. 1999;99:3181-3198
26. Betancourt T, Shah K, Brannon-Peppas L. Rhodamine-loaded poly(lactic-co-glycolic acid) nanoparticles for investigation of in vitro interactions with breast cancer cells. J Mater Sci Mater Med. 2009;20:387-395
27. Hubbell JA, Chilkoti A. Nanomaterials for drug delivery. Science. 2012;337:303-305
28. Lindegren S, Frost S, Bäck T, Haglund E, Elgqvist J, Jensen H. Direct Procedure for the Production of 211At-Labeled Antibodies with an ε-Lysyl-3-(Trimethylstannyl)Benzamide Immunoconjugate. J Nucl Med. 2008;49:1537-1545
29. Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicinconjugated PLGA-PEG block copolymer. J Control Release. 2000;70:63-70
30. Liang C, Yang Y, Ling Y, Huang Y, Li T, Li X. Improved therapeutic effect of folate-decorated PLGA-PEG nanoparticles for endometrial carcinoma. Bioorg Med Chem. 2011;19:4057-4066
31. Song Z, Feng R, Sun M, Guo C, Gao Y, Li L. et al. Curcumin-loaded PLGA-PEG-PLGA triblock copolymeric micelles: Preparation, pharmacokinetics and distribution in vivo. J Colloid Interface Sci. 2011;354:116-123
32. Wang S, Su R, Nie S, Sun M, Zhang J, Wu D. et al. Application of nanotechnology in improving bioavailability and bioactivity of diet-derived phytochemicals. J Nutr Bioche. 2014;25:363-376
33. Aguilar ZP. Types of Nanomaterials and Corresponding Methods of Synthesis. Nanomater Med Appl. 2013:33-82
34. Jensen AI, Binderup T, Kumar Ek P, Kjær A, Rasmussen PH, Andresen TL. Positron emission tomography based analysis of long-circulating cross-linked triblock polymeric micelles in a U87MG mouse xenograft model and comparison of DOTA and CB-TE2A as chelators of copper-64. Biomacromolecules. 2014;15:1625-1633
35. Jensen AI, Binderup T, Ek PK, Grandjean CE, Rasmussen PH, Kjær A. et al. PET imaging with copper-64 as a tool for real-time in vivo investigations of the necessity for cross-linking of polymeric micelles in nanomedicine. J Label Compd Radiopharm. 2017;60:366-374
36. Ebrahimnejad P, Dinarvand R, Jafari MR, Tabasi SAS, Atyabi F. Characterization, blood profile and biodistribution properties of surface modified PLGA nanoparticles of SN-38. Int J Pharm. 2011;406:122-127
37. Spetz J, Rudqvist N, Forssell-Aronsson E. Biodistribution and Dosimetry of Free 211At, 125I- and 131I- in Rats. Cancer Biother Radiopharm. 2013;28:657-664
38. Lundh C, Lindencrona U, Schmitt A, Nilsson M, Forssell-Aronsson E. Biodistribution of Free 211At and 125I- in Nude Mice Bearing Tumors Derived from Anaplastic Thyroid Carcinoma Cell Lines. Cancer Biother Radiopharm. 2007;21:591-600
39. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: The Phagocyte Problem Graphical Abstract HHS Public Access. Nano Today. 2015;10:487-510
40. Hadley SW, Wilbur DS, Gray MA, Atcher RW. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment using N-succinimidyl p-astatobenzoate: comparisons in vivo with the p-[125I]iodobenzoyl conjugate. Bioconjug Chem. 1991;2:171-179
41. Reist CJ, Foulon CF, Alston K, Bigner DD, Zalutsky MR. Astatine-211 labeling of internalizing anti-EGFRvIII monoclonal antibody using N-succinimidyl 5-[211At]astato-3-pyridinecarboxylate. Nucl Med Biol. 1999;26:405-411
42. Bäck TA, Jennbacken K, Hagberg Thulin M, Lindegren S, Jensen H, Olafsen T. et al. Targeted alpha therapy with astatine-211-labeled anti-PSCA A11 minibody shows antitumor efficacy in prostate cancer xenografts and bone microtumors. EJNMMI Res. 2020;10:1-12
43. Palm S, Bäck T, Claesson I, Danielsson A, Elgqvist J, Frost S. et al. Therapeutic Efficacy of Astatine-211-Labeled Trastuzumab on Radioresistant SKOV-3 Tumors in Nude Mice. Int J Radiat Oncol. 2007;69:572-579
44. Kato H, Huang X, Kadonaga Y, Katayama D, Ooe K, Shimoyama A. et al. Intratumoral administration of astatine-211-labeled gold nanoparticle for alpha therapy. J Nanobiotechnology. 2021;19:1-12
45. Lai TC, Cho H, Kwon GS. Reversibly core cross-linked polymeric micelles with pH- and reduction-sensitivities: effects of cross-linking degree on particle stability, drug release kinetics, and anti-tumor efficacy. Polym Chem. 2014;5:1650-1661
46. Lindegren S, Bäck T, Jensen HJ. Dry-distillation of astatine-211 from irradiated bismuth targets: a time-saving procedure with high recovery yields. Appl Radiat Isot. 2001;55:157-160
47. Jestin E, Bultel-Rivière K, Faivre-Chauvet A, Barbet J, Loussouarn A, Gestin JF. A simple and efficient method to label l-fucose. Tetrahedron Lett. 2006;47:6869-6873
48. Koziorowski J, Henssen C, Weinreich R. A new convenient route to radioiodinated N-succinimidyl 3- and 4-iodobenzoate, two reagents for radioiodination of proteins. Appl Radiat Isot. 1998;49:955-959
49. Garg PK, Archer GE, Bigner DD, Zalutsky MR. Synthesis of radioiodinated N-succinimidyl iodobenzoate: Optimization for use in antibody labelling. Int J Radiat Appl Instrumentation Part A Appl Radiat Isot. 1989;40:485-490
50. Coenen HH, Moerlein SM, StCklin G. No-Carrier-Added Radiohalogenation Methods with Heavy Halogens. Radiochim Acta. 1983;34:47-68
Author contact
![]() Corresponding authors: atijedk (A.I.J.); matthias.herthku.dk (M.M.H.)
Corresponding authors: atijedk (A.I.J.); matthias.herthku.dk (M.M.H.)