- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Imaging immune response
Imaging checkpoint molecules
Imaging engineered T cells
Imaging the tumor stroma
Imaging using nanomaterials
Conclusion
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2022; 6(3):286-305. doi:10.7150/ntno.66556 This issue Cite
Review
Molecular Immune Targeted Imaging of Tumor Microenvironment
Taha Rakhshandehroo, PhD1, Bryan Ronain Smith, PhD2, Hannah J. Glockner, PhD1, Mohammad Rashidian, PhD1, Neeta Pandit-Taskar, MD3 ![]()
1. Department of Imaging, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
2. Department of Biomedical Engineering, Michigan State University, East Lansing, MI, USA
3. Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY and Weill Cornell Medical College, New York, NY, USA
Received 2021-8-27; Accepted 2021-12-28; Published 2022-2-15
Abstract

Novel targeted therapies are rapidly emerging for the treatment of cancer. With the advent of new immune targeting agents, understanding the changes in the tumor microenvironment (TME) is critical. Given the complexity and several cellular mechanisms and factors that play a role in the TME, novel imaging methods to assess and evaluate the dynamic changes in the TME during treatment are needed. Several techniques are being developed for imaging TME including optical, fluorescence and photoacoustic methods. Positron emission tomography (PET) imaging can be used to track the dynamics of different molecular targets in the TME in live animals and in humans. Several novel PET imaging probes including radiolabeled antibodies, antibody fragments, and small molecules have been developed with many more that are under development preclinically and in early human studies. This review is a brief overview of some of the PET agents that are either in the preclinical developmental phase or undergoing early clinical studies.
Keywords: Biomarker, radiolabeled antibody, checkpoint inhibitors, PD-1, PD-L1, FDG
Introduction
Immune-directed therapies of malignancies are a growing strategy for treatment of multiple tumors. Current treatment strategies face challenges in clinical management, mainly related to variable responses and outcomes across different malignancies and patients. Several physiological factors and processes of the tumor microenvironment such as changes in extracellular matrix components, vascular or lymphatic structures in tumor, cellular alterations, stromal cell derived proteases or physiologic processes such as hypoxia, interstitial pressure and pH play a complex role in tumor growth and response to treatment [1,2]. Assessment of tumor microenvironment (TME) is critical for prognostication and optimizing the choice of targeting agent and dynamic evaluation of TME can help assess treatment effectiveness [3].
Noninvasive imaging plays a major role in the management of patients with cancer. Several imaging methods are currently being clinically utilized that provide unique information and allow for imaging multiple processes in tumor microenvironment. These broadly include imaging modalities such as optical imaging, functional imaging with positron emission tomography (PET) or single-photon emission tomography (SPECT), magnetic resonance imaging/spectroscopy (MRI/MRS), and ultrasound imaging. An imaging modality that accurately provides predictive and prognostic information is lacking and there is a critical unmet need for novel prognostic imaging agents. Additionally, given the complexity of the TME, monitoring dynamic changes during treatment and predicting outcomes is a challenge. Functional PET imaging with 18F-fluorodeoxyglucose (18F-FDG) has proven useful not only as a sensitive tool to detect and stage cancer, but also as a biomarker for prediction and prognostication in the treatment of malignancies and is currently the most widely used PET imaging radiotracer for management of malignancies [4]. However, 18F-FDG is an analog of glucose and is taken up by metabolically active glucose-avid cells expressing the GLUT1 and/or GLUT3 transporters [5]. Thus, besides cancer cells, activated immune cells in tumor or those associated with inflammation and physiologically glucose-avid tissues such as the brain and heart as well as inflammatory cells can thus uptake 18F-FDG. 18F-FDG therefore is not specific and has no ability to delineate subpopulations of cells, thereby limiting its power to evaluate TME and outcomes for immune-directed therapies. Novel targeted imaging agents are needed in order to detect and differentiate subclasses of cells within the TME.
Several antibody-based therapies exploiting tumor, immune, or vascular markers have been developed as cancer therapeutics. Such targeted agents can be useful to noninvasively monitor the composition of the TME, using functional imaging. Preclinical and early clinical studies to image subpopulations of immune cells, vasculature, presence or absence of cytokines, and to determine the suitability of a therapeutic strategy such as bi-specifics or CAR-T cell therapies are being conducted. Several imaging approaches generally utilize targeting agents such as antibody, antibody fragment, peptide or ligand, or other small molecules such as aptamer. Other techniques include reporter molecules that can be targeted for functional imaging using nanoparticles, fluorophores, or radionuclides. While several imaging techniques are being used, this review focuses primarily on functional imaging using radioisotopes and includes a brief discussion on nanoparticles.
Imaging using radiolabeled small molecules or antibodies
Small molecules, such as radiolabeled sugars, hormones, drugs, and nucleosides are the most prevalent form of radiolabeled agents for PET imaging. An advantageous attribute of these small molecules is the ability to permeate cell membranes and, therefore, access targets intracellularly. Additionally, due to their small size, these tracers rapidly clear from circulation and have higher tissue penetrance and permeation compared to antibody-based tracers.
Despite the favorable attributes of small molecules as imaging agents, they pose challenges as imaging agents to assess the immune response to cancer. Due to rapid metabolism and excretion, there can be high uptake in the kidneys and bladder.
Antibody-based PET imaging platforms allow for highly specific targeted imaging of immune processes. As compared to small molecules, the antibodies are larger molecules (~150 kDa) that may limit tissue penetration; the Fc region of antibodies binds to neonatal Fc receptor (FcRn), recycling the antibody back into circulation and protecting it from lysosomal degradation that also results in longer circulation. However, the advantage of antibodies is that they best resemble the characteristics of an antibody-drug. Smaller antibody fragments such as minibodies, cys-diabodies, single-chain Fvs (scFvs), and nanobodies have been developed and used for PET imaging. Antibody-fragments as imaging agents have combined advantage of smaller size resulting in more rapid clearance and higher tissue penetration as compared to antibodies.
Imaging immune response
In order to target immune cells and differentiate them from cancer cells, small molecule radiotracers have been developed targeting specific metabolic pathways that are upregulated in activated immune cells (Table 1). Most tissues rely on the de novo DNA synthesis pathway to produce nucleotides to support cellular growth and division. However, rapidly expanding immune cells utilize the nucleoside salvage pathway for DNA synthesis [6]. Deoxycytidine kinase (dCK) is highly expressed in lymphocytes and is the rate-limiting enzyme in the nucleoside salvage pathway [7]. 1-(2'-deoxy-2'-[18F]fluoroarabinofuranosyl) cytosine (18F-FAC), a nucleoside analog that is specific towards dCK, shows specific labeling of proliferating CD8+ T cells [8]. Preclinical studies indicated high signal-to-noise in lymphoid organs and bone marrow while ex vivo analysis illustrated high uptake in T cells, B cells, and CD11b+ myeloid cells. However 18F-FAC is likely not a suitable imaging tool for ultimate clinical translation because it is rapidly catabolized by cytidine deaminase [9].
Radiolabeled molecules or peptides for imaging
| Agent | Target | Class | Stage | Active clinical trials | References |
|---|---|---|---|---|---|
| 18F- AraG | Deoxyguanosine kinase (dGK) | Small Molecule | Clinical | NCT04052412, NCT03142204, NCT04186988, NCT03129061, NCT03684655, NCT03367962 | [10], [11], [135] |
| 18F-CFA | dCK | Small Molecule | Clinical | NCT03409419 | [136] |
| 1-L-18F-FETrp | IDO | Small Molecule | Preclinical | [137] | |
| 18F-FAC | Deoxycytidine kinase (dCK) | Small Molecule | Clinical | [8] | |
| 68Ga-WL12 | PD-L1 PD-L1 PD-L1 PD-L1 PD-L1 PD-L1 | Peptide | Preclinical | [138] | |
| 18F-FPy-WL12 | Peptide | Preclinical | [24] | ||
| 18F-NOTA-ZPD-L1_1 | Affibody | Preclinical | [139] | ||
| 64Cu-DOTA-FN3hPD-L1 | Adnectin | Preclinical | [140] | ||
| 68Ga-NOTA-Nb109 | Nanobody | Preclinical | [141] | ||
| 64Cu-DOTA-HAC-PD1 | High-affinity PD-1 ectodomain | Preclinical | [142], [143] |
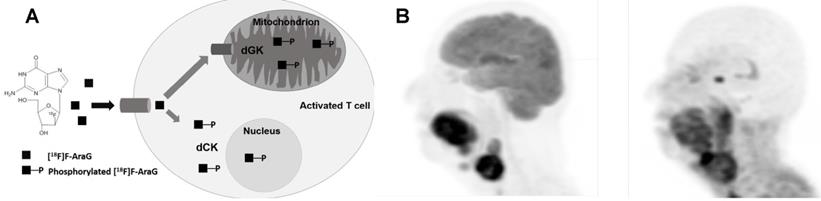
To overcome this issue, 9-(β-D-Arabinofuranosyl) guanine (AraG), a non-catabolizible imaging agent, was developed. As a guanosine analog, it can be phosphorylated by dCK and, consequently, trapped intracellularly in T cells in vivo without any toxicity [10, 11], therefore serving as an ideal targeted small molecule PET imaging tracer (Figure 1). In a murine model of colorectal cancer, two days post-administration of anti-PD-1 treatment, responder mice showed a significantly higher 18F-AraG PET signal in their tumors relative to a non-responder mice, indicating 18F-AraG PET was able to successfully predict the response to programmed death-1 (PD-1) blockade [11]. In patients, 18F-AraG preferentially accumulates in activated CD8+ T cells with about 7 times higher uptake in activated T cells as compared to murine cells [10]. Currently, four phase I (NCT04524195, NCT04052412, NCT04678440, NCT04186988) and two phase II (NCT04726215, NCT04260256) human studies are underway to study biodistribution and kinetics in patients with non-small cell lung cancer (NSCLC) treated with checkpoint blockade.
(A) Mechanism of uptake of 18F-ARAG: Reproduced from Levi J et al. CCR (doi:10.1158/0008-5472.CAN-19-0267). (B) Patient with head and neck carcinoma. PET/CT imaging with 18F-FDG and 18F-ARAG shows prominent uptake in the lesions in larynx; uptake is also noted in 18F-ARAG scan with heterogenous uptake. (Courtesy: Drs. Colevas, Sunwoo, and Davidzon, Stanford University) (Reprinted with permission from Elsevier, Pandit-Taskar N, Postow MA. Immune-Directed Molecular Imaging Biomarkers. Semin Nucl Med. 2020 Nov;50(6):584-603. doi: 10.1053/j.semnuclmed.2020.06.005. Epub 2020 Jul 15. PMID: 33059826.)
Although 18F-AraG can target expanding immune cells, it is not a representative readout for regions of T cell activity and killing. Targeted small molecules that bind to markers of T cell activity, either as released molecules or expressed on the cell surface, can be used to assess the T cell response to cancer. Granzyme B is a serine protease found in the granules of cytotoxic T cells and has been recognized as an important downstream effector of tumoral cytotoxic T cells. Granzyme B, upon secretion into the target cell through perforin-mediated pores, can cleave and activate caspase 8 and 10, leading to a signaling cascade that triggers apoptosis of the targe cell. Therefore, imaging granzyme B is expected to distinguish an active immune response from exhausted T cells. Granzyme B also stimulates the release of cytokines, can induce inflammation, and also is involved in extracellular matrix remodeling. Therefore, it has gained interest as a potential biomarker to predict the efficacy of immunotherapies [12]. A 68Ga-radiolabeled granzyme B-binding peptide (GZP) has been designed from the cleavage sequence of murine granzyme B, which has been modified to promote irreversible binding [13]. In a colorectal preclinical cancer model, tumors that responded to anti-CTLA-4 and anti-PD-1 treatment and imaged 1 hour post-injection of the tracer showed high uptake in the tumor while non-responders showed low tumor uptake [13]. Furthermore, in tumor models treated with a combination of checkpoint inhibitors, 68Ga-NOTA-GZP tracer uptake in the treatment group linearly correlated with percent response across all therapies, demonstrating its value as a predictive marker [14]. A first in-human phase I clinical imaging study (NCT04169321) to assess safety and kinetic of 68Ga-NOTA-hGZP is being conducted in 20 patients with melanoma or non-small cell lung cancer treated with pembrolizumab.
Antibodies or fragments have been developed to image myeloid cells that play key roles in shaping the immune status of the TME [15-17], [18]. Imaging their presence and activation status can help to better assess the response to immunotherapy. Tumor-associated macrophages can significantly influence the TME immune landscape by secreting different cytokines. M1-like macrophages secrete inflammatory cytokines, such as CCL5, CXCL9, and CXCL10, which can recruit and activate T cells, whereas M2-like macrophages secrete cytokines that repel T cells. Thus, imaging the presence and phenotypic status of macrophages in the TME can provide valuable information for the assessment of immunotherapy response. A 99mTc-labeled nanobody targeting the macrophage mannose receptor (MMR, CD206), a marker highly expressed by the immunosuppressive M2-like macrophages, has been utilized for SPECT imaging in preclinical models of lung and mammary tumors. This imaging agent was able to detect MMR+ macrophages in hypoxic regions of the tumor with clarity three hours post-injection [18]. Further preclinical PET imaging studies utilizing a human/mouse MMR cross-reactive 18F-labeled nanobody in a mouse model of lung cancer validated MMR+-macrophage imaging, suggesting it can serve as a suitable imaging tool for ultimate clinical translation [19].
CD11b is a myeloid cell marker and has served as a target for PET imaging of myeloid cells. An anti-CD11b 99mTc-labelled antibody was used in a mouse colon cancer model and showed high uptake in tumors six hours post-injection, indicating the presence of myeloid cells [20]. Similarly, an anti-CD11b nanobody was used to image tumor infiltration of myeloid cells in a melanoma model and could detect the tumor with clarity. Interestingly, penetration of CD11b+ cells to the tumor core was not found to be a negative predictor of tumor response to PD-1 blockade [21], potentially due to the diversity and polarization of CD11b+ cells, which can serve as both anti-tumor and pro-tumor roles.
Professional antigen presenting cells, including macrophages, B cells, and dendritic cells, are pivotal to development of an anti-tumor immune response. These cells are unique in their expression of MHC-II, which is used to present tumor antigens to CD4+ T cells. A nanobody against mouse MHC-II was developed and used for in vivo detection of tumor infiltrating MHC-II+ cells in both syngeneic and xenogeneic preclinical models. The 18F-radiolabaled nanobody could detect MHC-II+ cells in the tumor two hours post-injection [21]. An anti-human MHC-II nanobody was developed and used to image MHC-II+ cells in a humanized mouse model of graft versus host disease (GvHD). Mice that developed GvHD showed high uptake of the anti-MHC-II tracer in the liver [22]. Performing single-cell RNA sequencing on tumor infiltrating immune cells obtained from a responder and a non-responder mouse treated with PD-1 blockade showed a significant increase in the expression profile of class II MHC molecules on tumor immune infiltrating cells in the responder animal [21], suggesting that imaging class II MHC can be used to evaluate a response to treatment. The higher expression is potentially due to higher presence of IFN-γ in the TME of the responding tumor. The ability to predict and prognosticate using this target is yet to be proven. Additionally, the lack of ability to distinguish between different APCs using MHC-II to target can be limiting.
As innate lymphoid cells, natural killer (NK) cells interact with and destroy tumors via different mechanisms than T cells. NK cell activation can represent anti-tumor activity. Antibodies to NKp30, an activation natural cytotoxicity receptor expressed on NK cells, were radiolabeled with 64Cu or 89Zr. These stable and specific radiolabeled antibodies were injected into several murine xenograft and adoptive cell transfer models of renal cell carcinoma and imaged via PET to visualize NK cell activation.
Imaging checkpoint molecules
Preclinical imaging
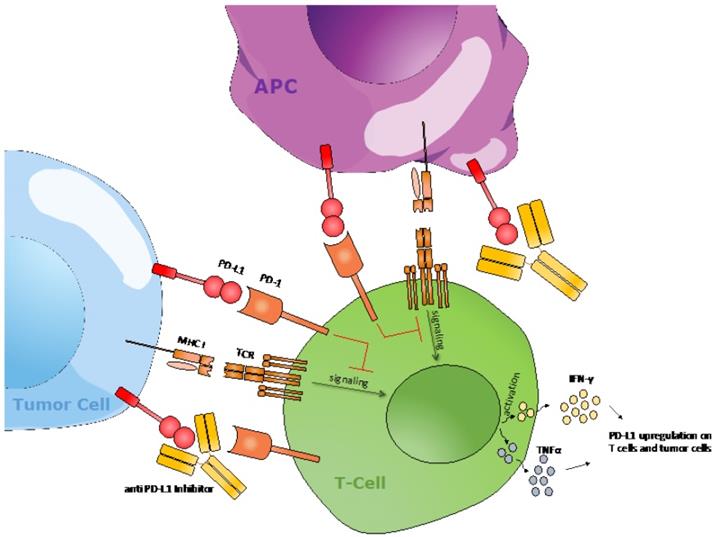
Checkpoint blockade treatments, such as anti-PD-1 and anti-CTLA-4 have shown impressive success in the treatment of several malignancies such as melanoma and non-small cell lung cancer (NSCLC). Checkpoint inhibitor therapies act by preventing immune suppression via temporarily blocking the interaction of checkpoint molecules with their receptors, resulting in continued activation of T cells that can lead to a more efficient anti-tumor immune response. Several studies have shown expression of PD-L1 is correlated to the response to PD-1 blockade (Figure 2) [23]. However, currently, the only clinically viable method to assess tumor expression of PD-L1 protein is through tumor biopsies, which is invasive, susceptible to sampling errors, practically restrictive, and lacks the ability to capture the heterogeneity of a tumor. Therefore, several approaches have been developed to noninvasively image PD-L1.
PD-1/PD-L1 checkpoint molecules regulate T cell activation. Upon peptide-MHC interaction with its T cell receptor in concurrence with costimulatory signals, T cells are activated, resulting in T cell-mediated killing and the release of cytokines, such as IFN-γ and TNFα. The interaction between PD-1 and PD-L1 results in the downregulation of anti-tumor T cell signaling. An agonistic anti-PD-(L)1 antibody will disrupt this interaction and subsequently help with T cell activation.
A cyclic 14-residue peptide, named WL12, that binds to PD-L1 with low nanomolar affinity (IC50 ~ 26-32 nM) was developed and used for PET imaging of PD-L1 expression. In mice bearing cancer xenografts, 18F-WL12 showed high specificity and affinity for PD-L1. The specific uptake of 18F-WL12 in tumors was shown to be reduced when cold peptide was injected prior to imaging, further showing the specificity of the observed signal. The biodistribution analysis of 18F-WL12 showed prominent uptake in hepatic, renal, and normal tissues. High tumor uptake was observed 2 hours post injection with %ID/g ranging from 7.16-8.86 at 1 and 2 hours post-injection [24]. A phase I clinical study in patients with solid tumors is planned (NCT04304066).
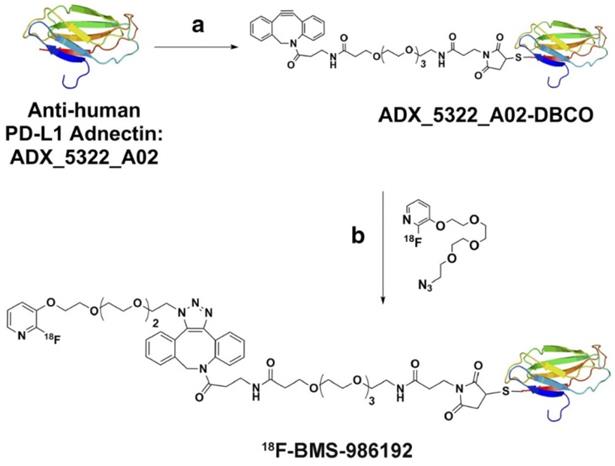
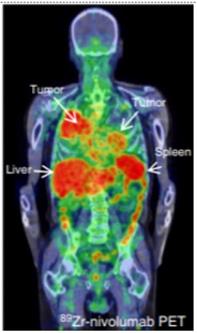
Adnectins, small protein molecules of ~10kDa size, have been developed against different targets as imaging agents. Adnectins are engineered based on the framework of the human 10th fibronectin type III domain (10FN3). They are structurally similar to variable domains of antibodies and are suitable as PET imaging agents owing to their small size, a non- or low-immunogenic profile, and pharmacokinetic properties [25]. Adnectins lack disulfide bonds and can be designed against a specific target with low nanomolar or even picomolar affinities. 18F-BMS-986192, an adnectin specific for PD-L1, was developed and used for imaging PD-L1 in lung cancer patients (Figure 3). The biodistribution showed uptake in marrow and spleen, sites of high PD-L1 expression, while uptake in lesions showed heterogeneity between patients as well as for different lesions in a patient. 89Zr-Nivolumab is radiolabeled an anti-PD-1 antibody that has shown ability to assess target expression in patients (Figure 4). Combined imaging with 89Zr-Nivolumab and 18F-BMS-986192 allowed for simultaneous assessment of both PD-1 and PD-L1, that demonstrated variation in uptake, thereby expression of PD-1 and PD-L1 across lesions in patients [26, 27]. Preclinical imaging in mice noted 3.5 times higher uptake in tumors expressing PD-L1 as compared to control animals. Specificity was further confirmed with blocking of radioligand binding in a dose-dependent manner [28].
Antihuman PD-L1 adnectin. Structure and synthesis of 18 F-BMS986192 antiPD-L1 adnectin. (Adapted from a figure that was originally published in JNM. Donnelly D et al. Synthesis and Biologic Evaluation of a Novel 18 F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J Nucl Med. 2018;59(3):529-535. © SNMMI.)
89Zr-labeled Nivolumab PET (37.09 MBq, 162 h p.i.) demonstrate heterogeneous tracer uptake within and between tumors. (Adapted for use under Creative Commons license http://creativecommons.org/licenses/by/4.0/ from Niemeijer, A.N., Leung, D., Huisman, M.C. et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat Commun 9, 4664 (2018). https://doi.org/10.1038/s41467-018-07131-y)
Clinical imaging
Immune checkpoint inhibitor therapy is rapidly becoming the mainstay in the management of several malignancies. Over the last several years, many radiolabeled antibodies targeting the checkpoint molecules have been developed and some are already in early clinical assessment (Figure 5).
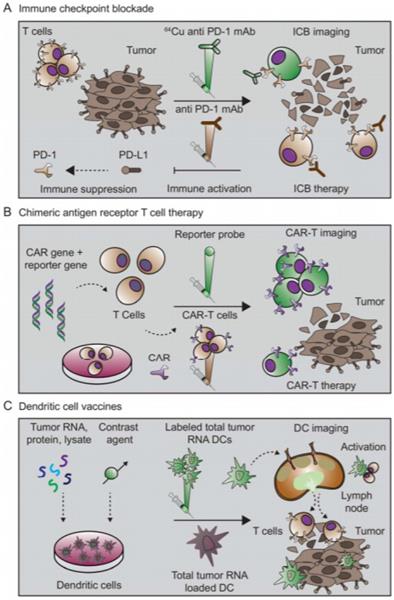
Immuno-imaging applications toward immunotherapy. Immune checkpoint blockade, CAR-Ts, and therapeutic vaccines constitute three important classes of cancer immunotherapy. (A) Immune checkpoints that regulate anti-tumor immunity have now been identified as promising therapeutic targets. Blocking signaling pathways that suppress anti-tumor immune responses has proven especially effective. In one approach, an anti-PD-1 mAb targets the PD-1 receptor on T cells, blocking ligation of the receptor and immunosuppression by PD-L1 on tumor cells. Anti-PD1 mAb administration thus leads to immune activation and therapeutic response. Imaging the expression of PD1 with a radiolabeled mAb may assist with selection of patients for treatment, optimal dosing, and response monitoring. (B) CAR-T strategies engineer a patient's immune cells ex vivo to express a receptor that can bind specifically to tumor cells. During this engineering process, a reporter gene can also be inserted to enable longitudinal tracking of the CAR-T cells. Upon administration, CAR-Ts seek out and destroy malignant tumor cells. Subsequent imaging with a reporter probe can give insights into their location and functional status. (C) Cancer vaccine strategies come in many formulations. In one approach, dendritic cells are pulsed with tumor antigen, lysate, or RNA. Dendritic cells then express tumor antigens on their MHC molecules, which are capable of eliciting a T cell-driven immune response. Successful responses require homing of dendritic cells to the lymph nodes and tumor. At these sites, the dendritic cells are capable of activating tumor-specific T cells. Labeling the dendritic cells with a contrast agent allows for assessment of successful homing of dendritic cells to lymph nodes and other secondary lymphoid sites. This knowledge can be utilized to inform both dose and route of vaccine administration. (Adapted from a figure that was originally published in JNM. Mayer AT and Gambhir SS. The Immunoimaging Toolbox. J Nucl Med. 2018;59(8):1174-1182. © SNMMI.)
Pembrolizumab and Nivolumab, FDA-approved monoclonal (IgG4) antibodies for targeting PD-1 receptors, have been radiolabeled with 64Cu or 89Zr for imaging (Table 2). Preclinical assessment with 89Zr-Pembrolizumab showed specific targeting of T cells in the spleen and in salivary and lacrimal glands, confirmed by tissue staining. The retained activity in tissue was long, with a liver clearance half-life of 168 hours [29]. Distribution of 89Zr-Nivolumab in healthy primates showed similar distribution with prominent uptake in liver, spleen, and lymph nodes, sites rich in T cell infiltration, confirmed with blocking of uptake with cold Nivolumab and histology analyses [30, 31]. Low uptake was noted in other tissues such as brain, heart, lung, kidney, and muscle, while the nodal uptake increased with time, suggesting continuous binding to T cell and accumulation. Atezolizumab and Durvalumab antibodies target PD-L1 and are FDA-approved for the treatment of certain malignancies, either as a single agent or in combination with other agents. Imaging with 89Zr-atezolizumab in 22 patients with either NSCLC, triple-negative breast cancer, or bladder cancer showed good targeting of tissues and lesions with PD-L1 expression, as correlated with immunohistochemistry [32]. There was prominent splenic, nodal, and bone marrow uptake and correlation of uptake to CD8 expression was noted in spleen though some uptake was also noted in some sites of inflammation. Tumor uptake was heterogenous and varied between tumor types and sites of lesions (Figure 6). Lower uptake was noted in breast cancer lesions compared to bladder or lung cancer and in bone lesions compared to liver lesions. Higher 89Zr-atezolizumab uptake in lesions was noted in those with better response and 89Zr-atezolizumab uptake showed better correlation than with immunohistochemistry or RNA-sequencing-based predictive biomarkers [32]. 89Zr-atezolizumab imaging may be useful to stratify and identify patients with RCC who are most likely to benefit from PD-1/PD-L1-directed treatment [33]. 89Zr-Durvalumab imaging trials in head and neck cancer and NSCLC undergoing treatment with durvalumab are underway [34] (NCT03853187 and NCT03829007).
Radiolabeled antibodies/antibody fragments/ligands for imaging
| Agent | Target | Type | Stage | Clinical trials | References |
|---|---|---|---|---|---|
| 89 Zr-Df-IAB22M2C | CD8 | Minibody | Clinical | NCT03107663 (completed) NCT03802123 | [84] |
| 64Cu-NOTA-ipilimumab-F(ab')2 | CTLA-4 | F(ab')2 | Preclinical | [144] | |
| 64Cu-NOTA-ipilimumab | Antibody IgG1 | Preclinical | [144] | ||
| 64Cu-DOTA-ipilimumab | Antibody IgG1 | Preclinical | [145] | ||
| 89Zr-ipilimumab | Antibody IgG1 | Clinical | NCT03313323, 2012-003616-31 | ||
| 18F-FB-anti-MMR 3.49 | Macrophage Mannose Receptor (MMR) | Nanobody | Preclinical | [19] | |
| 64Cu- VHH4 | MHC II | Nanobody | Preclinical | [22] | |
| 64Cu-NOTA-avelumab Fab | PD-L1 | Fab | Preclinical | [146] | |
| 111In-labeled atezolizumab | Antibody IgG1 | Preclinical | [147] | ||
| 64Cu-DOTA-atezolizumab | Antibody IgG1 | Preclinical | [147], [148] | ||
| 111In-PD-L1.3.1 | Antibody IgG1 | Preclinical | [149] | ||
| 89Zr-DFO-C4 | Antibody IgG1 | Preclinical | [150] | ||
| 64Cu-WL12 | Peptide | Clinical | NCT04304066 | [151], [152] | |
| 99mTc-NM-01 | Nanobody | Clinical | NCT02978196 (completed) | ||
| 89Zr-envafolimab | Nanobody Fc fusion | Clinical | NCT03638804 | [153], [154] | |
| 18F-BMS-986192 | Adnectin | Clinical | 2015-004760-11(completed) NCT03520634, NCT03843515, NCT03564197, NCT03843515, 2018-002643-28, | [27], [155] | |
| 89Zr-durvalumab | Antibody IgG1 | Clinical | 2015-005765-23, NCT03829007, NCT03853187 | [34] | |
| 89Zr-labeled avelumab | Antibody IgG1 | Clinical | NCT03514719 | [156] | |
| 89Zr-labeled atezolizumab | Antibody IgG1 | Clinical | NCT03850028, NCT04006522, NCT04222426, NCT02478099, 2019-001197-28, 2017-003511-20 | [32] | |
| 89Zr-CX-072 | Pro-antibody | Clinical | 2016-002490-36 | [157] | |
| 64Cu-pembrolizumab | PD-1 | Antibody IgG4 | Preclinical | [158], [159] | |
| 89Zr-pembrolizumab | Antibody IgG4 | Clinical | NCT02760225, NCT03065764, NCT03446911*, 2015-004260-10, 2016-003819-36 | [158], [29] | |
| 89Zr-nivolumab | Antibody IgG4 | Clinical | [27], [30] | ||
| 89Zr-DFO-AN-18 | IFN - γ | Intact antibody | Preclinical | [113] | |
| 64Cu-DOTA-etanercept | TNF-α | Anti-TNF-α drug (Etanercept) | Preclinical | [114] | |
| 18F-FB-IL-2 | IL-2 receptor | Labeled cytokine (IL-2) | Clinical | NCT03304223, NCT04163094, NCT02478099 | [117] |
| 68Ga-Ga-NODAGA-IL2 | Labeled cytokine (IL-2) | Preclinical | [122] | ||
| 18F-AlF-RESCA-IL2) | Labeled cytokine (IL-2) | Preclinical | [122] [160], [161] | ||
| 89Zr-Bevacizumab | VEGF | Intact antibody | Clinical | NCT01081613, NCT01338090, NCT01894451, NCT01028638, NCT00991978, NCT00831857, NCT00970970 |
*PET imaging using radiotracer is considered a secondary outcome
Radiolabeled 89Zr-atezolizumab imaging in patients showing heterogenous uptake within lesions in the thorax: (left) mediastinal lesion in a patient with lung cancer, (middle) abdominal wall metastatic bladder cancer, and (right) node in abdomen and metastatic bone lesion from breast cancer (arrows). (Adapted from Bensch E et al. Nat Med. 2018 Dec;24(12):1852-1858.)
Lymphocyte-activation gene 3 (LAG-3) is a cell surface marker predominantly expressed in CD3-positive T cells with low expression in normal or malignant B cells, NK cells, and myeloid cells [35] and in PD-1-positive CD8+ and CD4+ tumor-infiltrating lymphocytes. LAG-3 negatively regulates T cell proliferation and activation with a role in suppressing the active T cells [36]. 89Zr-REGN3767 is a radiolabeled antibody against LAG-3 antibody that is being evaluated in a first-in-human study in patients with lymphoma (NCT04566978) and other advanced malignancies (NCT03005782).
Imaging engineered T cells
Preclinical imaging
Chimeric antigen receptor (CAR) T cells have shown great promise in treatment of blood-borne malignancies. CAR is a synthetic receptor capable of binding to a target and eliciting an activation and expansion signal in T cells such that it, ultimately, causes the killing of the target cell (Figure 7). Currently, CAR T cells specific for CD19 have been FDA-approved for the treatment of various B cell malignancies such as large B cell lymphoma or acute lymphoblastic leukemia (ALL) [37, 38], while BCMA CAR T cells were recently approved for the treatment of multiple myeloma [39]. Despite these encouraging results, patients face detrimental side effects, such as cytokine release syndrome [40] or B cell aplasia [41, 42]. Therefore, visualizing and imaging the behavior and dynamics of CAR T cells in vivo can become a robust clinical tool to assess or even predict response to CAR T cell treatment, for which several strategies have been developed (Table 3).
Reporter gene imaging T-cells
| Agent | Reporter gene | Species | Type | Stage | |
|---|---|---|---|---|---|
| 18F-FHBG | Herpes Simplex Virus - Thymidine Kinase (HSV-TK) | Herpes Simplex Virus 1 | Enzyme | Clinical | [162], [163] |
| 18F-DCFPyL | Glutamate carboxypeptidase 2 (PSMA) | Human | Cell surface enzyme | Preclinical | [58] |
| 99mTcO4- | Sodium Iodine Symporter (NIS) | Human | Transporter | Preclinical | [49] |
| 123I-MIBG, 124I-MIBG | Norepinephrine Transporter (NET) | Human | Cell surface receptor | Preclinical | [50] |
| 68Ga-DOTATOC | Somatostatin Receptor 2 (SSTR2) | Human | Cell surface receptor | Preclinical | [51] |
| 18F-TMP | Dihydrofolate reductase enzyme (DHFR) | Escherichia coli | Enzyme | Preclinical | [164] |
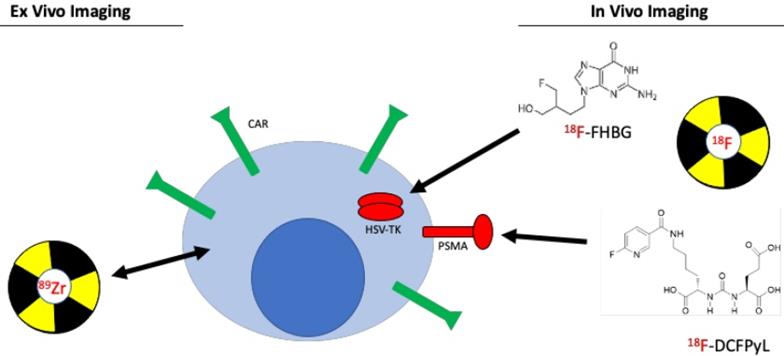
Imaging chimeric antigen receptor (CAR) T cells. Two different approaches are commonly used to image engineered T cells. Ex vivo labeling involves direct use of the PET radiotracer on the engineered T cell, either through binding of the tracer to proteins on the plasma membrane or through passive transport across the plasma membrane. This method is simple and easy to conduct, however, due to effluxion over time as well as dilution due to T cell expansion, same-day imaging is required. In vivo labeling is a much more robust and utilized imaging method. The method utilizes a small molecule PET radiotracer specific to a reporter gene that is co-expressed with the chimeric antigen receptor. This approach allows for longitudinal assessment of CAR T cells.
Direct ex vivo labeling of immune cells isolated from patients with a radioactive tracer after being transduced and just before being adoptively transferred allows the CAR T cells to be monitored longitudinally by PET imaging over time in vivo. This strategy enables simple, rapid, and specific labeling. However, the first instance of ex vivo labeling of human immune cells with 99mTc found only 50% cellular viability post-labeling [43]. Therefore, ionophore chelators, such as 8-hydroxyquinoline (oxine) or hexamethylpropyleneamine (HMPAO), which carry radioisotopes across the plasma membrane, were developed to increase labeling efficiency and viability. Utilizing these ionophores revealed 98% viability with 73% and 44% labeling efficiency when using 111In-oxine and 99mTc-HMPAO, respectively [44].
A preclinical study used 89Zr-oxine to label anti-IL-13R2 CAR T cells and found labeling efficiency was 75% while labeled cells retained more than 60% of the 89Zr after 6 days in vitro [45]. The labeled CAR T cells were delivered intraventricularly to mice bearing patient-derived glioblastoma multiforme (GBM) cancer cells. The CAR T cells were detectable by PET at least 6 days post-injection within intracranial tumors with no effect on CAR T cell-mediated tumor killing [45]. These results indicate that ex vivo labeling maintains CAR T cell function and PET tracer signal in vivo throughout a relevant timeframe.
Ex vivo labeling is a simple technique to label immune cells. Because CAR T cells are transduced and expanded ex vivo, the ex vivo labeling approach can be a simple addition to a clinical protocol. Although, upon infusion, the expansion of CAR T cells will dilute the radiotracer among newly expanded cells causing a decrease in signal. Therefore, imaging must be conducted in a short timeframe after infusion. Furthermore, decay and effluxion of the radioisotope also necessitate immediate PET imaging upon CAR T cell infusion. Therefore, other methods, such as in vivo imaging, should be utilized that require less restricting imaging needs. In particular, while long-term cell labeling typically requires introduction of a reporter gene, shorter-term labeling can be accomplished by in vivo immune cell labeling or ex vivo cell labeling and re-injection [46].
In order to overcome the shortcomings of ex vivo labeling, imaging CAR T cells using a reporter gene can be used. A reporter gene expressed in CAR T cells allows for imaging in vivo using a radioactive small molecule specific for the reporter. This strategy allows for imaging despite migration, homing, and expansion of the CAR T cell, making imaging of cell trafficking at any time point possible in vivo. Herpes simplex virus thymidine kinase (HSV-TK), human norepinephrine transporter (hNET), and sodium-iodide symporter (hNIS), among others, are established reporter genes that have been used to image CAR T cells [47]. HSV-TK, however, can serve not only as a PET reporter imaging system, but also in a bifunctional role a potential suicide switch for CAR-T cells upon infusion of a drug such as ganciclovir [48].
The HSV-TK has been widely used as a PET reporter gene system. Upon infusion of the CAR T cells expressing HSV-TK, 18F-labeled Penciclovir, a small molecule inhibitor for HSV-TK, can be administered, which will target the CAR T cells. The advantage of this reporter gene is the specificity of the small molecule and the ability to inject the targeted imaging agent at any timepoint. This system shows high specificity and low background, especially when using a mutant HSV-TK with higher affinity for Penciclovir. However, a concern that exists with the HSV-TK reporter system is a potential immune response against the reporter. Therefore, human reporter genes, such as sodium iodide symporter (hNIS) [49], norepinephrine transporter (hNET) [50], and somatostatin receptor 2 (SSTR2) [51] have been used to label CAR T cells. Each of these human reporter systems have certain advantages and disadvantages. For example, the advantage of hNIS is that it is non-immunogenic, not internalized, and is not expressed in necrotic cells [52]. However, the hNIS reporter system has high background because hNIS is expressed in many normal epithelial tissues and carcinomas [53]. In contrast, SSTR2 human reporter gene has an advantage because of its low baseline expression in normal tissue as well as available clinically approved specific radiotracers, such as 68Ga octreotide analogues (68Ga-DOTATOC) [54]. However, SSTR2 is not as sensitive due to internalization upon interaction with ligands [55].
PET imaging employing split reporter strategies has also been used to evaluate protein-protein interactions in vivo based on protein-fragment complementarity. The basis of this approach is that splitting a reporter protein into two fragments abolishes function, but when the fragments are brought within close proximity, reporter activity is partially restored [56]. When parts of the reporter protein are split between two interacting proteins, the dynamic localization of their interactions can thus be visualized. Using HSV-TK, PET imaging can visualize the interactions. An early example of this approach visualized protein-protein interactions of several protein partners, such as hypoxia-inducible factor-1α (HIF-1α) and the von Hippel-Lindau tumor suppressor, with strong potential to be used in monitoring immune cell therapies [57].
A recent preclinical study utilized PSMA as a reporter gene for noninvasive imaging of CAR T cells [58]. PSMA is a cell surface protein whose expression is limited to the prostate gland making it an ideal reporter candidate as a reporter [59]. A radiolabeled small molecule, 2-(3-(1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl)-ureido)-pentanedioic acid (18F-DCFPyL), was developed as a highly sensitive and specific probe targeting PSMA [60]. CAR T cells co-expressing anti-CD19 CAR and PSMA were detected with high sensitivity in vitro and in vivo while CAR T cell infiltration into primary and metastatic Nalm6 tumors was visualized [58]. Interestingly, response to CAR T cell therapy was not correlated with detection of CAR T cells in the blood using flow cytometry, which is the current clinical practice. However, CAR T cell infiltration visualized by PET was correlated to response, indicating that noninvasive imaging of CAR T cells may be a more clinically relevant prognostic tool.
While endogenous biomarkers are typically attractive for imaging cancer, the sensitivity and/or specificity is often not sufficient to drive clinical adoption and success. Recently, macrophages were engineered as immunological sensors that generate a synthetic bioluminescent reporter when they polarize toward a more M2-like tumor-associated macrophage profile [61]. This strategy was accomplished by pairing luciferase expression to the activation of the immunosuppressive arginase-1 promoter. In cancer models, once the macrophages trafficked to tumor sites, arginase-1 was activated, simultaneously releasing luciferase for detection by bioluminescence as a site-specific indicator of M2-polarization.
These preclinical studies illustrate the utility of in vivo labeling to track and monitor CAR T cells and macrophages. Both ex vivo and in vivo labeling approaches provide advantages including simplicity or temporal control, respectively, as well as drawbacks, such as a time-limited protocol or potential immunogenicity, respectively. However, in vivo imaging techniques using reporter genes provide the ideal characteristics for clinical practice due to the freedom to image at any timeframe after treatment. Reporter genes, such as PSMA and 18F-DCFPyL, allow monitoring of CAR T cell trafficking to and infiltration into the tumor and metastatic sites while providing better prognostic power compared to serial assessment in the peripheral blood. In conclusion, PET imaging of CAR T cells using reporter systems is highly translatable in the clinic and shows potential as a prognostic tool.
Another strategy of significant interest is imaging infiltration of tumors by T cells. Cytotoxic CD8+ T cells directly mediate the anti-tumor immune response and, thus, are an important target for noninvasive imaging [62]. Similarly, CD4+ T cells play an essential role in shaping the immune landscape of the TME and visualizing their presence in the tumor holds prognostic value [63]. Several strategies have been developed to image T cells in both preclinical and clinical settings. This includes using antibody and antibody fragments targeting CD3, CD4 and CD8, markers of T, helper T and cytotoxic T cells, respectively [64]. A radiolabeled murine anti-CD3 antibody was found to be able to detect tumor infiltrating lymphocytes in a syngeneic murine model 72 hours post-injection [65]. In vivo imaging using a 89Zr-radiolabeled anti-CD4 cys-diabody resulted in high uptake in lymphoid organs 20 h post-injection [66]. 89Zr-radiolabeled minibodies specific for mouse CD8 developed for PET imaging of CD8+ T cells resulted in high uptake in the lymphoid organs in mice 4 hours post-injection [67]. These preclinical studies demonstrate that in vivo imaging of T cells is feasible.
Other preclinical studies were able to correlate the T cell infiltration into tumors with treatment outcomes. A 89Zr-labeled anti-CD3 antibody used in a preclinical colorectal cancer model found that tumors that responded to anti-CTLA-4 treatment had higher T cell infiltration compared to non-responders [68]. A cys-diabody against murine CD8 molecule was developed for detection of tumor-infiltrating CD8+ lymphocytes [69]. In a murine model of colorectal cancer, mice were treated with an anti-PD-1 antibody and imaged with the radiolabeled cys-diabody 48 hours later. Animals that received the treatment were found to have greater infiltration of CD8+ T cells into the tumors, indicating the predictive power of CD8+ PET imaging in response to anti-PD-1 treatment [69]. A nanobody against murine CD8 was developed and used to detect CD8+ T cells in vivo. Addition of a 20 kDa polyethylene glycol (PEG) moiety to the nanobody significantly enhanced signal-to-background ratio and reduced uptake in the kidneys, which was attributed to its increased circulatory half-life and hydrophilicity [70]. Homogeneous distribution of CD8+ T cells in tumors was found to be a predictive marker of response to CTLA-4 blockade. In a B16 melanoma model, mice treated with CTLA-4 blockade and GVAX vaccination [71] were monitored longitudinally by CD8+ PET imaging once a week for four weeks. The 89Zr-labelled PEGylated anti-CD8 nanobody was similarly used to image T cell infiltration in a murine colorectal cancer model treated with PD-1 blockade [21]. Responder animals showed a significant increase of T cell infiltration into the tumors, whereas T-cells in non-responders remained mostly around the tumor periphery [21]. These studies illustrate that infiltration of T cells into tumors is a predictive response to immunotherapy and can easily be translated into the clinic.
While imaging of infiltrating T cells using anti-CD3 or anti-CD8 antibodies has prognostic value, detected T cells may be exhausted or anergic and not capable of actively mounting an anti-tumor response. An important attribute of activated T cells is their increased surface expression of costimulatory molecules such as ICOS, 4-1BB, and OX40 [72-74]. Therefore, noninvasively imaging these costimulatory molecules can provide a better indication of T cell activation. A recent study using a 89Zr-labeled anti-ICOS antibody in a lung cancer model detected activated CD4+ and CD8+ T cells in the tumor and tumor-draining lymph nodes in response to a STING immune agonist treatment [75]. In a similar study, a radiolabeled anti-OX40 antibody was used to detect activated T cells in A20 B cell leukemia tumors and tumor draining lymph nodes 4, 16, and 24 hours after tracer administration in response to CpG vaccination [76]. These results show that imaging activated CD8+ T cells is a valuable tool for assessing the immune response to cancer immunotherapy.
Clinical imaging of T cells
The presence of activated cytotoxic CD8+ T cells are important for initiating and mediating a successful response to CTLA-4 and PD-1/PD-L1 checkpoint blockade treatments [77-79] (Figure 5) and has been shown to be of prognostic value for tumor response in patients [80]. Imaging of CD8+ cells is, therefore, an attractive approach for assessment of the anti-tumor status of TME in patients.
89Zr-IAB22M2C, a radiolabeled 80 kDa anti-CD8 minibody, is currently being evaluated in a phase II multicenter study (NCT03802123). IAB22M2C is an engineered bivalent homodimer with each monomer consisting of a single-chain variable fragment (scFv) linked to the human IgG1 CH3 domain from the humanized heavy and light chain sequences of murine anti-human OKT8 antibody. The minibody is humanized, biologically inert, and does not interact with the FcRn recycling receptor [67, 69, 81, 82]. Preclinical studies of IAB22M2C and desferrioxamine -conjugated (Df)-IAB22M2C showed retention of high-affinity binding to human T cells (binding EC50 = 0.4 nM) and HPBALL leukemia cells and no measurable impact on proliferation or depletion of CD8+ T cells with incubation with peripheral blood mononuclear cells (PBMCs) from healthy human donors [83]. No acute effects of intravenous administration of Df-IAB22M2C were noted on CD8+ T cell populations or cytokine release in humanized NSG mice engrafted with CD34+ stem cells.
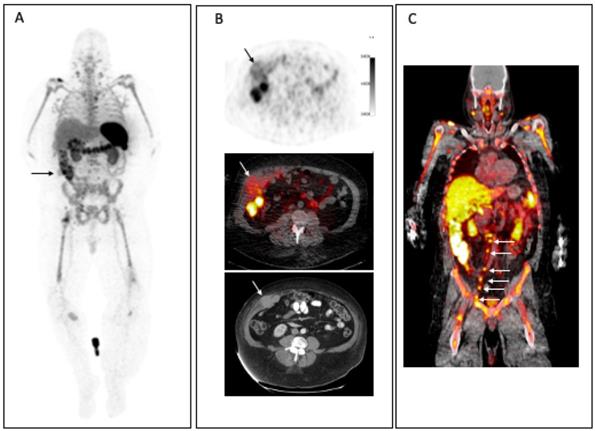
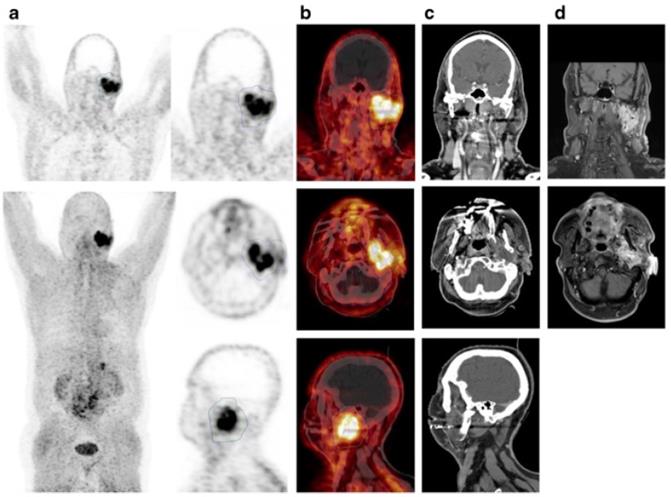
A phase I first-in-human study with 89Zr-Df-IAB22M2C PET imaging showed specific targeting of CD8+ T cell-rich tissue in patients with multiple solid tumors that were either on treatment with checkpoint inhibitors or planned to be treated with checkpoint inhibitors [84]. Prominent uptake in bone marrow, spleen, and lymph nodes at 6-24 h post-injection suggested selective accumulation in CD8+ T cell-rich tissues (Figure 8). 89Zr-IAB22M2C was noted in lesions in two patients that were being treated with immune checkpoint inhibitor therapy, suggesting possible enhanced modulation of TILs and presence of higher concentration of CD8+ T cells visualized by 89Zr-Df-IAB22M2C PET/CT imaging, which was confirmed by biopsy correlation that showed CD8+ T cell infiltration by immunohistochemistry in 89Zr-Df-IAB22M2C-positive lesion. In one patient who had not received immunotherapy, lesions were negative on 89Zr-IAB22M2C imaging, possibly suggesting a lack of TIL stimulation and infiltration. 89Zr-IAB22M2C-negative tumors in three patients with metastatic lung cancer lacked 18F-FDG uptake on concurrent PET imaging suggesting either lack of active disease or chronic low TIL infiltration [84]. A phase II multicenter study evaluating 89Zr-IAB22M2C imaging at baseline prior to and after checkpoint blockade treatment on patients with multiple solid tumors is underway (NCT03802123). In this study, 89Zr-IAB22M2C imaging and uptake is to be correlated with biopsies. Another CD8-targeting antibody for PET imaging, 89Zr-ZED88082A, is being evaluated in a phase I trial (NCT04029181, NCT02478099).
Patient with metastatic melanoma. (A) MIP images at 24h. p.i 3.0 mCi 89Zr-Df-IAB22M2C demonstrates prominent uptake in spleen, bone marrow and nodes, which are sites of T cell-rich tissue. Multiple focal areas of CD8 tracer uptake are noted including right flank muscle metastasis (arrow), lymph nodes in the neck, chest, pelvis, and groin and soft tissue in right lower extremity. (B) CD8+ tracer uptake is seen in the corresponding right flank muscle metastasis on axial CD8+ PET, fused CD8 PET/CT images and diagnostic CT. (C) Coronal PET/CT fusion images highlight the pelvic and retroperitoneal CD8+ nodal uptake (arrows).
Antibody or antibody fragments specific for CD3, a general marker for all T cells, and CD4, a marker for helper T cells, are being developed for clinical translation. 89Zr-labeled GK1.5 cDb is a radiolabeled diabody targeting CD4 that has shown T cell infiltration related uptake in spleen, blood, lymph nodes, and thymus [66]. A novel approach uses bispecific T-cell engager 89Zr-AMG 211 directed against tumor carcinoembryonic antigen (CEA) and CD3 on T-cells. In a small clinical study in 9 patients with advanced gastrointestinal adenocarcinomas, imaging showed uptake in CD3-rich tissue such as spleen and bone marrow [85]. Although these approaches are in the early phases of developments, they may allow for imaging and tracking of T cells in real time.
Imaging the tumor stroma
Imaging angiogenesis
Angiogenesis is the process by which new blood vessels are developed from a pre-existing vascular structure. This process can be induced by the tumor, due to the absence of oxygen and nutrients, and is important for tumor growth and metastasis. The regulation of angiogenesis involves many signaling pathways, receptors, and ligands such as αvβ3 integrin, platelet-derived growth factor (PDGF), matrix metalloproteinases (MMPs), hypoxia-inducible factor 1 (HIF-1), and vascular endothelial growth factor (VEGF). All these pathways are potential targets for therapies and can be considered as targets for imaging agents.
Considering these various angiogenesis-stimulating factors, VEGF is considered the most potent and predominant factor [86]. VEGF-A is the most prominent growth factor in the VEGF family and it stimulates angiogenesis in healthy and tumor tissue by signaling through VEGF receptor-2 [87]. Proteolytic enzymes in tumors may activate or release growth factors from the extracellular matrix (ECM) or act directly on the ECM itself, thereby facilitating angiogenesis or tumor cell migration. Bevacizumab is an FDA-approved monoclonal antibody that targets VEGF-A and is approved for the treatment of multiple cancer types, including colorectal, lung, breast, brain, and ovarian cancer. It has also been utilized as a PET imaging agent both preclinically and clinically. A preclinical PET imaging study found much higher tumor uptake of 89Zr-bevacizumab compared to 89Zr-IgG in nude mice implanted with the human ovarian cell line, SKOV-3, after 72 hours [88]. Ex vivo biodistribution studies after 168 hours showed high uptake of the radioisotope in the tumor, spleen, liver, and bone.
Additionally, a clinical PET imaging study conducted on renal cell carcinoma patients using 89Zr-bevacizumab as an imaging agent before and after treatment with bevacizumab with interferon-α or sunitinib found tumor uptake of the imaging agent was high yet heterogeneous in patients. Interestingly, treatment with bevacizumab in combination interferon-α strongly decreased tumor uptake of the imaging agent, indicating a correlation between response to treatment and tumor PET signal, whereas sunitinib treatment resulted in a modest decrease in uptake [89]. Similarly, a PET clinical study using 89Zr-bevacizumab in children with diffuse intrinsic pontine glioma (DIPG) found five of seven patients displayed PET signal at the tumor site with one patient showing signal at multiple metastatic sites [90].
Although VEGF-A is an important growth factor in the angiogenic process, its cognate receptor, VEGFR-2, is a major mediator of angiogenesis. VEGFR-2 is highly expressed in a range of solid tumors [91] and is associated with ovarian cancer progression [92]. Different approaches have been taken to image VEGFR-2, the earliest of which was the use of radiolabeled VEGF-A. A preclinical study utilizing the VEGF165 and VEGF121 growth factor, the active soluble secreted VEGF isoforms, radio iodinated with 123I found the radiolabeled agent can bind to human umbilical vein endothelial cells as well as a range of established tumor cell lines. This led to clinical studies utilizing 123I-VEGF165 and 123I-VEGF121 in patients with gastrointestinal tumors [93] and pancreatic lesions [94]. These proof-of-principle studies led to the use of VEGF as a PET imaging agent for the detection of VEGFR-2 expression. Nude mice implanted with U87 glioblastoma cells subjected to PET imaging using 64Cu-DOTA-VEGF121 were found to have high uptake of the radiotracer in small (high VEGFR-2 expression) tumors but low uptake in larger (low VEGFR-2 expression) tumors [95]. Unfortunately, high renal uptake with no observable clearance was also observed, likely due to high VEGFR-1 expression in the kidneys. To overcome this challenge, a VEGF121 mutant (VEGFDEE) was developed that is specific for VEGFR-2 only, thereby avoiding renal uptake and toxicity [96]. The study found high uptake of 64Cu-DOTA-VEGFDEE in VEGFR-2 expressing 4T1 tumors implanted in BALB/c but significantly lower renal uptake compared to 64Cu-DOTA-VEGF121. A more recent study utilized a 12-amino-acid peptide within exon 6 of VEGF-A (VEGF125-136) that was first identified as an inhibitor to VEGFR as a targeted PET imaging agent. They found 64Cu-DOTA-VEGF125-136 effectively targets B16F10 and U87 cells in vivo with great pharmacokinetic properties only showing noteworthy kidney and liver uptake in ex vivo biodistribution studies [97].
Imaging of fibroblasts
Cancer-associated fibroblasts (CAF) are one of the main cellular components in the TME and play a major role in regulating the behavior of tumors. CAFs are activated fibroblasts with a mesenchymal cell lineage that secrete a variety of soluble factors that facilitate the progression of cancer stemness, immune regulation, angiogenesis, drug resistance, extracellular matrix remodeling, and other biological processes [98], [99, 100]. Therefore, noninvasive imaging of CAFs can provide insight into the TME. Fibroblast activation protein (FAP) is a cell surface antigen of reactive tumor stromal fibroblasts found in epithelial cancers but not in normal tissues. More than 90% of lung, breast, and colon carcinomas have FAP expression in their stroma. Of note, FAP has been associated with poor prognosis. In patients with gastric cancer, upregulation of FAP was noted in poorly differentiated tumors and correlated with more adverse clinical and pathological characteristics including histology and pathological stage [101]. In a phase I clinical study, a FAP-specific antibody, F-19, was developed and used as an imaging agent. The radiolabeled antibody was used for detection of FAP in the tumor stroma in patients with colorectal carcinoma where hepatic metastasis was shown [102]. In another study, 131I-Sibrotuzumab, a humanized antibody against human FAP, was used for imaging in a phase I dose escalation study with cold Sibrotuzumab. Lesions were visualized with high contrast and no normal organ uptake was noted [103]. Subsequent studies have focused on the development of FAP as a target for cancer therapies [104, 105]. Furthermore, a 68Ga-radiolabeled FAP inhibitor ligand, 68Ga-FAPI (Figure 9), was shown to target tumor tissue in head and neck tumors with high signal-to-background ratio. Radiation planning volumes assessed with 68Ga-FAPI showed larger gross tumor volume as compared to conventional imaging [106]. Further developments for targeting FAP include fluorescence activatable bispecific endoglin-fibroblast, FAP-targeting liposomes [107], and adoptively transferred CAR-T cells directed to FAP [108].
PET imaging with 68Ga-FAPI is superior to conventional MRI scan. (A, B) A patient receiving a PET scan with 68Ga-FAPI to noninvasively image a mucoepidermoid carcinoma lesion on the left parotid gland. PET images show tracer uptake at the tumor site with low background. (C, D) Conventional MRI imaging shows diffuse tumor infiltration, thereby making delineation of tumor and stromal tissue difficult and subjective. (Adapted for use under Creative Commons license http://creativecommons.org/licenses/by/4.0/ from Syed, M., Flechsig, P., Liermann, J., et al. Fibroblast activation protein inhibitor (FAPI) PET for diagnostics and advanced targeted radiotherapy in head and neck cancers. Eur J Nucl Med Mol Imaging 47, 2836-2845 (2020). doi: 10.1007/s00259-020-04859-y)
Imaging cytokines
Although mutations in oncogenes and tumor suppressor genes are the drivers of tumor progression, the TME plays a pivotal role in tumor growth and dynamic between the tumor and immune cells. Cytokines in the TME can contribute to both an anti-tumor immune stimulatory (for example IL-2 family, INF-α, IL-10, IL-12) and pro-tumor inflammatory response (such as TNF- α, TGF-β, IL-8) [109]. Cytokines present an anti-tumor response through increasing antigen-presentation, recruiting and enhancing the cytotoxicity of T cells [110]. However, pro-tumor cytokines promotes an anti-inflammatory response that induces an immunosuppressive tumor landscape [111, 112]. Therefore, the detection of cytokines in the TME can directly gauge the immune landscape of the tumor and provide a prognostic, and even predictive, tool for determining the response to immunotherapy.
Several preclinical studies have noninvasively imaged cytokines. For example, a 89Zr-labeled antibody has been developed to detect IFN-γ in a mammary cancer model [113]. Mice implanted with breast cancer cells expressing HER2 when treated with a HER2-specific DNA vaccine were found to have higher uptake of the tracer in tumors. Interestingly, tracer uptake was positively correlated with response to therapy, indicating that IFN-γ can be a prognostic imaging marker [113]. In another study, 64Cu was conjugated to Etanercept, a TNF Receptor 2 IgG1 Fc fusion protein that binds to and inhibits TNF-α, and used to noninvasively image TNF-α in a model of acute and chronic inflammation [114]. Inflammation was induced by injection with 12-O-tetradecanoyl-phorbol-13-acetate (TPA) into the right ear and the tracer was subsequently injected. Tracer uptake significantly increased in the right ear after only a single injection of TPA, illustrating the sensitivity of Etanercept as an imaging agent.
Another PET imaging strategy involves the use of radiolabeled cytokines to detect activated T cells or other immune cells expressing their cognate receptors. Several studies have used radiolabeled Interleukin-2 (IL-2), a 15 kD alpha-helical protein produced by activated T cells that supports differentiation in regulatory T-cells and proliferation in effector T cells, for detection of activated T cells expressing the high affinity IL-2 receptor, comprised of a tricomplex IL2 receptor alpha (IL2RA) (CD25), beta (IL2RB) and common gamma chain [109], [115-118]. A fluorinated IL-2, N-(4-[18F]fluorobenzoyl)-IL-2 (18F-FB-IL-2) has been used to detect activated T cells in the TME of a lung cancer model to monitor cancer therapy [119]. Mice whose tumors were treated with either radiation or vaccination showed greater uptake of 18F-FB-IL-2 in their tumors, indicating an increased presence of activated T cells. Labeling of IL2 with (99m)Tc bifunctional chelating agent succinimidyl-6-hydrazinopyridine-3-carboxylate (HYNIC-NHS) and tricine, as co-ligand has been developed for imaging of IL-2 to track T cells in patients [120]. It binds to the IL-2 tri- receptor complex, expressed on TILs, and following internalization, the Tc99m/18F moiety is imaged. Feasibility and safety assessment in patients was favorable with only minor grade I toxicities such as pain or pruritis noted. In clinical studies, 99mTc-HYNIC-IL-2 imaging pre- and post-12 weeks of immune checkpoint therapy in metastatic melanoma showed correlation between change in lesion size post-therapy with change in the tracer uptake [121]. A phase I clinical trial in melanoma patients treated with immune checkpoint inhibitors is underway (NCT02922283). 18F-labeled IL2 (N-(4-18F-fluorobenzoyl)interleukin-2 18F-FB-IL2) has been evaluated preclinically [116] and, more recently, 18F-AlF-RESCA-IL2 and 68Ga-Ga-NODAGA-IL2 have also been developed and evaluated preclinically [122].
Imaging using nanomaterials
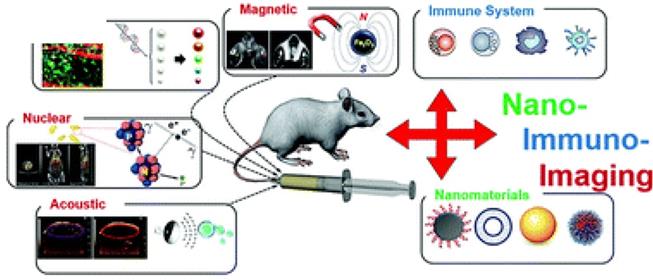
Nanomaterials have been used as contrast agents for in vivo imaging for several decades due to their capacity for high payload and avidity, robust targeting, and multimodal functionality [123, 124]. Contrast agent-containing nanomaterials moreover can be engineered to specifically bind/target immune cells and molecules in what is known as nano-immunoimaging, lying at the intersection of nanotechnology, imaging, and immunology [125, 126]. Nano-immunoimaging spans the existing imaging modalities, including magnetic, optical, acoustic, and nuclear approaches [127] (Figure 10). Here we describe molecularly targeted nanomaterials and intrinsically targeted nanomaterials.
Modalities for nano-immunoimaging. (Adapted from Wang P et al. NanoImmunoimaging, Issue 4, 2020.)
Molecularly targeted nanomaterials
Nanomaterials are ideal to increase imaging agent avidity due to their high surface area and surface to volume ratios, allowing many targeting ligands per nanoparticle [123]. Because tumor-associated macrophages (TAMs) can indicate cancer progression, the number of TAMs in a tissue is likely to impact immunotherapeutic response. A polyglucose nanoparticle (NP) was labeled with 64Cu to quantify TAMs in living subjects. Tumors high in TAM displayed >700% higher amounts of a model therapeutic NP versus TAM-deficient tumors based on PET, indicating that radiolabeled polyglucose NPs targeting TAM could be eventually used for patient stratification in immunotherapy trials [128]. Many other ligands conjugated to NPs may be employed in order to target immune cells, include those typically used in immune targeting strategies for nuclear medicine as described elsewhere in this review, e.g., antibodies/nanobodies/ diabodies, peptides, small molecules, and nucleic acids [129].
Intrinsically targeted nanomaterials
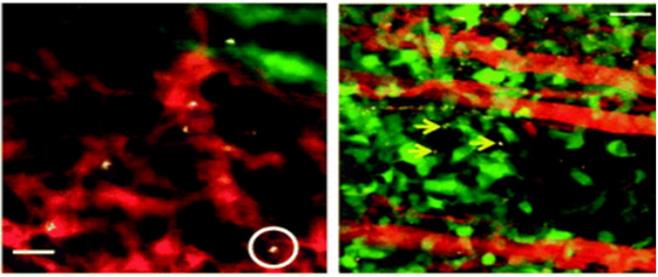
NPs can also display intrinsic cellular selectivity. We distinguish intrinsic selectivity from common properties, such as phagocytic uptake of many NP types by phagocytic cells (such as macrophages) that lead to uptake of many NP types. In contrast, NPs that are intrinsically selective will likely yield many benefits due to their capabilities to internalize in cell subsets other than macrophages, and to more simply inject the NPs in vivo without needing to harvest cells, isolate them, and then reinject them. In such ex vivo labeling cases, the cell phenotype could for instance change prior to reinjection, which adds risk as well as extra costs and time, making intrinsic in vivo labeling a highly attractive alternative to ex vivo labeling. The biggest challenge for in vivo labeling lies in endowing the capacity to target a particular subpopulation of cells with sufficient selectivity. This is true because most cell types must be identified by a set of multiple cell surface receptors (e.g., by flow cytometric analyses), not a single marker as would be required for typical in vivo targeting. While 100% targeting accuracy is not required to ensure effective delivery strategies in imaging (and therapy), it is clear that relatively high specificity is required alongside minimal off-target uptake. In vivo, exceptionally high intrinsic selectivity of single-walled carbon nanotubes (SWNTs) has been shown toward a subset of monocytes, inflammatory monocytes [130]. Approximately 100% of circulating inflammatory (Ly-6Chi) murine monocytes in the blood circulating took up SWNTs after injection, while under 3% of any other circulating cell type, including other myeloid cell subsets such as other monocytes, took them up. Visualization of these processes using intravital microscopy (IVM) exposed exceedingly quick SWNT uptake into inflammatory monocytes in the blood and ensuing trafficking into the tumor as a 'Trojan Horse' (Figure 11) as shown by both IVM and photoacoustic imaging [125, 130]. IVM is a powerful optical imaging tool that provides dynamic insights into biological and nanomaterial activities in living subjects at sub-micron resolution typically via fluorescence [131]. Another key advantage of IVM is its capability for multi-plexing many different colors, at least 5 at a time, which could be used to separately label and simultaneously track multiple different immune cell types within the same animal. Indeed, while this strategy selectively targets monocytes, one of the major challenges in the field is to boost the number of immune cell subsets (including valuable targets like cytotoxic T cells, NK, and NKT cells) that NPs can very selectively internalize within, and this high-reward objective in nano-immunoimaging is particularly important to decrease side effects from off-target binding that may lead to extraneous and difficult-to-interpret image signal [125].
SWNT uptake into circulating cells and deposition into tumor in a mouse model of glioblastoma multiforme (u87-mg cells) (left). Representative intravial fluorescence image of single walled carbon nanotube (SWNT)-laden circulating cells in tumor vasculature (one example cell is circled). (Adapted from Wang P et al. NanoImmunoimaging, Issue 4, 2020; originally reproduced by permission of Nature.)
Optical coherence tomography (OCT) is an in vivo imaging modality that applies light scattering within tissues to generate images, typically contrast-free. It boasts high resolution (only slightly worse than fluorescence) and deeper penetration capabilities than IVM. While one disadvantage of OCT is that its contrast agents are far less developed than other modalities, recent progress has produced scattering nanomaterials that add the ability to molecularly target or otherwise track cells using imaging contrast agents [131]. In one immune cell tracking application, macrophages and activated microglia in orthotopically implanted brain tumors naturally took up large gold nanorods, which produce substantial OCT contrast [132]. Given the ability to dynamically track these myeloid cells within tumors down to single-cell resolutions, it may be possible to closely monitor intratumoral macrophage responses to chemo- and immunotherapies in the future.
We note that nanomaterials do not necessarily need to be targeted, intrinsically or molecularly, if internalization is achieved ex vivo. For instance, immune cell subsets that have been harvested can be specifically isolated, and then embedded with imageable nanomaterials ex vivo, intended for reinjection and homing to the tumor microenvironment for imaging [46, 133].
Photoacoustic imaging (PAI) is an emerging imaging modality that benefits from many of the advantages of optical and ultrasonic imaging by sending sound 'in' (via laser) and creating tomographic images from sound 'out' (via ultrasonic transducers) to produce high spatial resolution images in fairly deep tissues (up to 7-8 cm) [123]. T cells labelled ex vivo with a photoacoustic dye were injected IV into mice bearing tumors. PAI was used as a cell tracking modality, able to assess T cell migration into tumors, with a peak at 12 hours at a depth of nearly 1 cm. Improvements to PAI immunoimaging approaches are likely, including development of PAI-active nanomaterial agents that target immune cells in vivo. For instance, SWNTs were used to target inflammatory monocytes to image inflammatory disease (atherosclerotic plaque) using PAI, and similar approaches are likely to enable PAI of cancer with utility in tracking the trafficking of inflammatory monocytes and macrophages in the TME [134].
Conclusion
As novel molecularly targeted therapeutic agents are becoming available for different malignancies, predicting the response and pharmacodynamic assessment of therapy is becoming more critical, particularly for immunotherapy. Assessing tumor target expression for treatment planning and management remains a clinical challenge. The complex nature of the TME and the processes that independently or interactively act in response to the changes with treatment and tumor growth require sophisticated and accurate methods to evaluate these changes. Several approaches, as mentioned in this review, are being explored to address these challenges. However, such methods are in the early phases of development, either clinically or preclinically. While many of these approaches are unique and hold promise for enabling a better understanding of changes in the tumor milieu, further work is needed for successful translation and implementation in clinical care.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591-6
2. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y. et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8:761-73
3. Perez-Romero K, Rodriguez RM, Amedei A, Barcelo-Coblijn G, Lopez DH. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. Int J Mol Sci. 2020 21
4. Gambhir SS, Czernin J, Schwimmer J, Silverman DH, Coleman RE, Phelps ME. A tabulated summary of the FDG PET literature. J Nucl Med. 2001;42:1S-93S
5. Mochizuki T, Tsukamoto E, Kuge Y, Kanegae K, Zhao S, Hikosaka K. et al. FDG uptake and glucose transporter subtype expressions in experimental tumor and inflammation models. J Nucl Med. 2001;42:1551-5
6. Griffith DA, Jarvis SM. Nucleoside and nucleobase transport systems of mammalian cells. Biochim Biophys Acta. 1996;1286:153-81
7. Arner ES, Flygar M, Bohman C, Wallstrom B, Eriksson S. Deoxycytidine kinase is constitutively expressed in human lymphocytes: consequences for compartmentation effects, unscheduled DNA synthesis, and viral replication in resting cells. Exp Cell Res. 1988;178:335-42
8. Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N. et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2'-deoxycytidine analog. Nat Med. 2008;14:783-8
9. Schwarzenberg J, Radu CG, Benz M, Fueger B, Tran AQ, Phelps ME. et al. Human biodistribution and radiation dosimetry of novel PET probes targeting the deoxyribonucleoside salvage pathway. Eur J Nucl Med Mol Imaging. 2011;38:711-21
10. Namavari M, Chang YF, Kusler B, Yaghoubi S, Mitchell BS, Gambhir SS. Synthesis of 2'-deoxy-2'-[18F]fluoro-9-beta-D-arabinofuranosylguanine: a novel agent for imaging T-cell activation with PET. Mol Imaging Biol. 2011;13:812-8
11. Levi J, Lam T, Goth SR, Yaghoubi S, Bates J, Ren G. et al. Imaging of Activated T Cells as an Early Predictor of Immune Response to Anti-PD-1 Therapy. Cancer Res. 2019;79:3455-65
12. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388-400
13. Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K. et al. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017;77:2318-27
14. Larimer BM, Bloch E, Nesti S, Austin EE, Wehrenberg-Klee E, Boland G. et al. The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin Cancer Res. 2019;25:1196-205
15. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB. et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839-49
16. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74:7250-9
17. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6:1670-90
18. Movahedi K, Schoonooghe S, Laoui D, Houbracken I, Waelput W, Breckpot K. et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 2012;72:4165-77
19. Blykers A, Schoonooghe S, Xavier C, D'Hoe K, Laoui D, D'Huyvetter M. et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J Nucl Med. 2015;56:1265-71
20. Cheng D, Zou W, Li X, Xiu Y, Tan H, Shi H. et al. Preparation and Evaluation of 99mTc-labeled anti-CD11b Antibody Targeting Inflammatory Microenvironment for Colon Cancer Imaging. Chem Biol Drug Des. 2015;85:696-701
21. Rashidian M, LaFleur MW, Verschoor VL, Dongre A, Zhang Y, Nguyen TH. et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc Natl Acad Sci U S A. 2019;116:16971-80
22. Van Elssen C, Rashidian M, Vrbanac V, Wucherpfennig KW, Habre ZE, Sticht J. et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J Nucl Med. 2017;58:1003-8
23. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X. et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129
24. Lesniak WG, Mease RC, Chatterjee S, Kumar D, Lisok A, Wharram B. et al. Development of [(18)F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol Imaging. 2019;18:1536012119852189
25. Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein engineering, design & selection: PEDS. 2011;24:3-9
26. Huisman M, Niemeijer AL, Windhorst B, Schuit R, Leung D, Hayes W. et al. Quantification of PD-L1 expression with [(18)F]BMS-986192 PET/CT in patients with advanced stage non-small-cell lung cancer. J Nucl Med. 2020
27. Niemeijer AN, Leung D, Huisman MC, Bahce I, Hoekstra OS, van Dongen G. et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat Commun. 2018;9:4664
28. Donnelly DJ, Smith RA, Morin P, Lipovsek D, Gokemeijer J, Cohen D. et al. Synthesis and Biologic Evaluation of a Novel (18)F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J Nucl Med. 2018;59:529-35
29. England CG, Ehlerding EB, Hernandez R, Rekoske BT, Graves SA, Sun H. et al. Preclinical Pharmacokinetics and Biodistribution Studies of 89Zr-Labeled Pembrolizumab. J Nucl Med. 2017;58:162-8
30. England CG, Jiang D, Ehlerding EB, Rekoske BT, Ellison PA, Hernandez R. et al. (89)Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer. Eur J Nucl Med Mol Imaging. 2018;45:110-20
31. Cole EL, Kim J, Donnelly DJ, Smith RA, Cohen D, Lafont V. et al. Radiosynthesis and preclinical PET evaluation of (89)Zr-nivolumab (BMS-936558) in healthy non-human primates. Bioorganic & medicinal chemistry. 2017;25:5407-14
32. Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC. et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. 2018;24:1852-8
33. Vento J, Mulgaonkar A, Woolford L, Nham K, Christie A, Bagrodia A. et al. PD-L1 detection using (89)Zr-atezolizumab immuno-PET in renal cell carcinoma tumorgrafts from a patient with favorable nivolumab response. J Immunother Cancer. 2019;7:144
34. Verhoeff S, Donk PPvd, Aarntzen EHJG, Miedema IHC, Oosting S, Voortman J. et al. 89Zr-durvalumab PD-L1 PET in recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck. Journal of Clinical Oncology. 2020;38:3573 -
35. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics. 1994;39:213-7
36. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917-27
37. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ. et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449-59
38. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O. et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377:2545-54
39. Mullard A. FDA approves first BCMA-targeted CAR-T cell therapy. Nature reviews Drug discovery. 2021;20:332
40. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25
41. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I. et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709-20
42. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099-102
43. Papierniak CK, Bourey RE, Kretschmer RR, Gotoff SP, Colombetti LG. Technetium-99m labeling of human monocytes for chemotactic studies. J Nucl Med. 1976;17:988-92
44. Becker W, Schomann E, Fischbach W, Borner W, Gruner KR. Comparison of 99Tcm-HMPAO and 111In-oxine labelled granulocytes in man: first clinical results. Nucl Med Commun. 1988;9:435-47
45. Weist MR, Starr R, Aguilar B, Chea J, Miles JK, Poku E. et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with (89)Zr-Oxine. J Nucl Med. 2018;59:1531-7
46. Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011;8:677-88
47. Yaghoubi SS, Campbell DO, Radu CG, Czernin J. Positron emission tomography reporter genes and reporter probes: gene and cell therapy applications. Theranostics. 2012;2:374-91
48. Murty S, Labanieh L, Murty T, Gowrishankar G, Haywood T, Alam IS. et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020;80:4731-40
49. Emami-Shahri N, Foster J, Kashani R, Gazinska P, Cook C, Sosabowski J. et al. Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat Commun. 2018;9:1081
50. Doubrovin MM, Doubrovina ES, Zanzonico P, Sadelain M, Larson SM, O'Reilly RJ. In vivo imaging and quantitation of adoptively transferred human antigen-specific T cells transduced to express a human norepinephrine transporter gene. Cancer Res. 2007;67:11959-69
51. Vedvyas Y, Shevlin E, Zaman M, Min IM, Amor-Coarasa A, Park S. et al. Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight. 2016;1:e90064
52. Penheiter AR, Russell SJ, Carlson SK. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr Gene Ther. 2012;12:33-47
53. Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K. et al. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab. 2003;88:1880-8
54. Zhang H, Moroz MA, Serganova I, Ku T, Huang R, Vider J. et al. Imaging expression of the human somatostatin receptor subtype-2 reporter gene with 68Ga-DOTATOC. J Nucl Med. 2011;52:123-31
55. Barsegian V, Hueben C, Mueller SP, Poeppel TD, Horn PA, Bockisch A. et al. Impairment of lymphocyte function following yttrium-90 DOTATOC therapy. Cancer Immunol Immunother. 2015;64:755-64
56. Paulmurugan R, Umezawa Y, Gambhir SS. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci U S A. 2002;99:15608-13
57. Massoud TF, Paulmurugan R, Gambhir SS. A molecularly engineered split reporter for imaging protein-protein interactions with positron emission tomography. Nat Med. 2010;16:921-6
58. Minn I, Huss DJ, Ahn HH, Chinn TM, Park A, Jones J. et al. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci Adv. 2019;5:eaaw5096
59. Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81-5
60. Szabo Z, Mena E, Rowe SP, Plyku D, Nidal R, Eisenberger MA. et al. Initial Evaluation of [(18)F]DCFPyL for Prostate-Specific Membrane Antigen (PSMA)-Targeted PET Imaging of Prostate Cancer. Mol Imaging Biol. 2015;17:565-74
61. Aalipour A, Chuang HY, Murty S, D'Souza AL, Park SM, Gulati GS. et al. Engineered immune cells as highly sensitive cancer diagnostics. Nat Biotechnol. 2019;37:531-9
62. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ. et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400-4
63. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM. et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696-701
64. Wei W, Jiang D, Ehlerding EB, Luo Q, Cai W. Noninvasive PET Imaging of T cells. Trends Cancer. 2018;4:359-73
65. Beckford Vera DR, Smith CC, Bixby LM, Glatt DM, Dunn SS, Saito R. et al. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS One. 2018;13:e0193832
66. Freise AC, Zettlitz KA, Salazar FB, Lu X, Tavare R, Wu AM. ImmunoPET Imaging of Murine CD4(+) T Cells Using Anti-CD4 Cys-Diabody: Effects of Protein Dose on T Cell Function and Imaging. Mol Imaging Biol. 2017;19:599-609
67. Tavare R, McCracken MN, Zettlitz KA, Knowles SM, Salazar FB, Olafsen T. et al. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 2014;111:1108-13
68. Larimer BM, Wehrenberg-Klee E, Caraballo A, Mahmood U. Quantitative CD3 PET Imaging Predicts Tumor Growth Response to Anti-CTLA-4 Therapy. J Nucl Med. 2016;57:1607-11
69. Tavare R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB. et al. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res. 2016;76:73-82
70. Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C. et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. 2017;214:2243-55
71. Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K. et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539-43
72. Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S. et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity. 2000;13:95-105
73. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445-55
74. DeBenedette MA, Shahinian A, Mak TW, Watts TH. Costimulation of CD28- T lymphocytes by 4-1BB ligand. J Immunol. 1997;158:551-9
75. Xiao Z, Mayer AT, Nobashi TW, Gambhir SS. ICOS Is an Indicator of T-cell-Mediated Response to Cancer Immunotherapy. Cancer Res. 2020;80:3023-32
76. Alam IS, Mayer AT, Sagiv-Barfi I, Wang K, Vermesh O, Czerwinski DK. et al. Imaging activated T cells predicts response to cancer vaccines. J Clin Invest. 2018;128:2569-80
77. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW. et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol. 2012;30:2678-83
78. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
79. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O. et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109-19 e10
80. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011;105:93-103
81. Knowles SM, Wu AM. Advances in immuno-positron emission tomography: antibodies for molecular imaging in oncology. J Clin Oncol. 2012;30:3884-92
82. Olafsen T, Sirk SJ, Olma S, Shen CK, Wu AM. ImmunoPET using engineered antibody fragments: fluorine-18 labeled diabodies for same-day imaging. Tumour Biol. 2012;33:669-77
83. Tavare R, McCracken MN, Zettlitz KA, Salazar FB, Olafsen T, Witte ON. et al. Immuno-PET of Murine T Cell Reconstitution Postadoptive Stem Cell Transplantation Using Anti-CD4 and Anti-CD8 Cys-Diabodies. J Nucl Med. 2015;56:1258-64
84. Pandit-Taskar N, Postow MA, Hellmann MD, Harding JJ, Barker CA, O'Donoghue JA. et al. First-in-Humans Imaging with (89)Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J Nucl Med. 2020;61:512-9
85. Moek KL, Waaijer SJH, Kok IC, Suurs FV, Brouwers AH, Menke-van der Houven van Oordt CW. et al. (89)Zr-labeled Bispecific T-cell Engager AMG 211 PET Shows AMG 211 Accumulation in CD3-rich Tissues and Clear, Heterogeneous Tumor Uptake. Clin Cancer Res. 2019;25:3517-27
86. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
87. Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw. 2009;20:158-63
88. Nagengast WB, Vries EGd, Hospers GA, Mulder NH, Jong JRd, Hollema H. et al. In vivo VEGF Imaging with Radiolabeled Bevacizumab in a Human Ovarian Tumor Xenograft. Journal of Nuclear Medicine. 2007;48:1313-9
89. Oosting SF, Brouwers AH, van Es SC, Nagengast WB, Oude Munnink TH, Lub-de Hooge MN. et al. 89Zr-bevacizumab PET visualizes heterogeneous tracer accumulation in tumor lesions of renal cell carcinoma patients and differential effects of antiangiogenic treatment. J Nucl Med. 2015;56:63-9
90. Jansen MH, Veldhuijzen van Zanten SEM, van Vuurden DG, Huisman MC, Vugts DJ, Hoekstra OS. et al. Molecular Drug Imaging: (89)Zr-Bevacizumab PET in Children with Diffuse Intrinsic Pontine Glioma. J Nucl Med. 2017;58:711-6
91. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581-611
92. Rudlowski C, Pickart AK, Fuhljahn C, Friepoertner T, Schlehe B, Biesterfeld S. et al. Prognostic significance of vascular endothelial growth factor expression in ovarian cancer patients: a long-term follow-up. Int J Gynecol Cancer. 2006;16(Suppl 1):183-9
93. Li S, Peck-Radosavljevic M, Koller E, Koller F, Kaserer K, Kreil A. et al. Characterization of (123)I-vascular endothelial growth factor-binding sites expressed on human tumour cells: possible implication for tumour scintigraphy. Int J Cancer. 2001;91:789-96
94. Li S, Peck-Radosavljevic M, Kienast O, Preitfellner J, Havlik E, Schima W. et al. Iodine-123-vascular endothelial growth factor-165 (123I-VEGF165). Biodistribution, safety and radiation dosimetry in patients with pancreatic carcinoma. Q J Nucl Med Mol Imaging. 2004;48:198-206
95. Cai W, Chen K, Mohamedali KA, Cao Q, Gambhir SS, Rosenblum MG. et al. PET of vascular endothelial growth factor receptor expression. J Nucl Med. 2006;47:2048-56
96. Wang H, Cai W, Chen K, Li ZB, Kashefi A, He L. et al. A new PET tracer specific for vascular endothelial growth factor receptor 2. Eur J Nucl Med Mol Imaging. 2007;34:2001-10
97. Hu K, Shang J, Xie L, Hanyu M, Zhang Y, Yang Z. et al. PET Imaging of VEGFR with a Novel (64)Cu-Labeled Peptide. ACS Omega. 2020;5:8508-14
98. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nature reviews Drug discovery. 2019;18:99-115
99. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. 2019;18:70
100. Su S, Chen J, Yao H, Liu J, Yu S, Lao L. et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell. 2018;172:841-56 e16
101. Liu J, Huang C, Peng C, Xu F, Li Y, Yutaka Y. et al. Stromal fibroblast activation protein alpha promotes gastric cancer progression via epithelial-mesenchymal transition through Wnt/ beta-catenin pathway. BMC Cancer. 2018;18:1099
102. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M. et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol. 1994;12:1193-203
103. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W. et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639-47
104. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613-25
105. Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J Natl Cancer Inst. 2012;104:1320-34
106. Syed M, Flechsig P, Liermann J, Windisch P, Staudinger F, Akbaba S. et al. Fibroblast activation protein inhibitor (FAPI) PET for diagnostics and advanced targeted radiotherapy in head and neck cancers. Eur J Nucl Med Mol Imaging. 2020;47:2836-45
107. Tansi FL, Ruger R, Kollmeier AM, Rabenhold M, Steiniger F, Kontermann RE. et al. Targeting the Tumor Microenvironment with Fluorescence-Activatable Bispecific Endoglin/Fibroblast Activation Protein Targeting Liposomes. Pharmaceutics. 2020 12
108. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154-66
109. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL. et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120:6-15
110. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11-22
111. von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Johnk C. et al. Systemic and local immunosuppression in pancreatic cancer patients. Clin Cancer Res. 2001;7:925s-32s
112. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. 2007;44:2850-9
113. Gibson HM, McKnight BN, Malysa A, Dyson G, Wiesend WN, McCarthy CE. et al. IFNgamma PET Imaging as a Predictive Tool for Monitoring Response to Tumor Immunotherapy. Cancer Res. 2018;78:5706-17
114. Cao Q, Cai W, Li ZB, Chen K, He L, Li HC. et al. PET imaging of acute and chronic inflammation in living mice. Eur J Nucl Med Mol Imaging. 2007;34:1832-42
115. Di Gialleonardo V, Signore A, Glaudemans AW, Dierckx RA, De Vries EF. N-(4-18F-fluorobenzoyl)interleukin-2 for PET of human-activated T lymphocytes. J Nucl Med. 2012;53:679-86
116. van der Veen EL, Antunes IF, Maarsingh P, Hessels-Scheper J, Zijlma R, Boersma HH. et al. Clinical-grade N-(4-[(18)F]fluorobenzoyl)-interleukin-2 for PET imaging of activated T-cells in humans. EJNMMI Radiopharm Chem. 2019;4:15
117. Hartimath SV, Draghiciu O, van de Wall S, Manuelli V, Dierckx RA, Nijman HW. et al. Noninvasive monitoring of cancer therapy induced activated T cells using [(18)F]FB-IL-2 PET imaging. Oncoimmunology. 2017;6:e1248014
118. van der Veen EL, Suurs FV, Cleeren F, Bormans G, Elsinga PH, Hospers GAP. et al. Development and Evaluation of Interleukin-2-Derived Radiotracers for PET Imaging of T Cells in Mice. J Nucl Med. 2020;61:1355-60
119. Hartimath SV, Khayum MA, van Waarde A, Dierckx R, de Vries EFJ. N-[(11)C]Methyl-AMD3465 PET as a Tool for In vivo Measurement of Chemokine Receptor 4 (CXCR4) Occupancy by Therapeutic Drugs. Mol Imaging Biol. 2017;19:570-7
120. D'Alessandria C, di Gialleonardo V, Chianelli M, Mather SJ, de Vries EF, Scopinaro F. et al. Synthesis and optimization of the labeling procedure of 99mTc-HYNIC-interleukin-2 for in vivo imaging of activated T lymphocytes. Mol Imaging Biol. 2010;12:539-46
121. Markovic SN, Galli F, Suman VJ, Nevala WK, Paulsen AM, Hung JC. et al. Non-invasive visualization of tumor infiltrating lymphocytes in patients with metastatic melanoma undergoing immune checkpoint inhibitor therapy: a pilot study. Oncotarget. 2018;9:30268-78
122. van der Veen EL, Suurs FV, Cleeren F, Bormans G, Elsinga PH, Hospers GAP. et al. Development and evaluation of interleukin-2 derived radiotracers for PET imaging of T-cells in mice. J Nucl Med. 2020
123. Smith BR, Gambhir SS. Nanomaterials for In vivo Imaging. Chem Rev. 2017;117:901-86
124. Smith BR. Nanotherapeutics for cardiovascular disease. Nat Rev Cardiol. 2021;18:617-8
125. Wang P, Kim T, Harada M, Contag C, Huang X, Smith BR. Nano-immunoimaging. Nanoscale Horiz. 2020;5:628-53
126. Zhu X, Li J, Peng P, Hosseini Nassab N, Smith BR. Quantitative Drug Release Monitoring in Tumors of Living Subjects by Magnetic Particle Imaging Nanocomposite. Nano Lett. 2019;19:6725-33
127. Ma X, Zhao Y, Liang XJ. Theranostic nanoparticles engineered for clinic and pharmaceutics. Accounts of chemical research. 2011;44:1114-22
128. Kim HY, Li R, Ng TSC, Courties G, Rodell CB, Prytyskach M. et al. Quantitative Imaging of Tumor-Associated Macrophages and Their Response to Therapy Using (64)Cu-Labeled Macrin. ACS Nano. 2018;12:12015-29
129. Mayer AT, Gambhir SS. The Immunoimaging Toolbox. J Nucl Med. 2018;59:1174-82
130. De la Zerda A, Zavaleta C, Keren S, Vaithilingam S, Bodapati S, Liu Z. et al. Carbon nanotubes as photoacoustic molecular imaging agents in living mice. Nature nanotechnology. 2008;3:557-62
131. Si P, Honkala A, de la Zerda A, Smith BR. Optical Microscopy and Coherence Tomography of Cancer in Living Subjects. Trends Cancer. 2020;6:205-22
132. SoRelle ED, Yecies DW, Liba O, Bennett FC, Graef CM, Dutta R. et al. Spatiotemporal Tracking of Brain-Tumor-Associated Myeloid Cells in vivo through Optical Coherence Tomography with Plasmonic Labeling and Speckle Modulation. ACS Nano. 2019;13:7985-95
133. Jung KO, Kim TJ, Yu JH, Rhee S, Zhao W, Ha B. et al. Whole-body tracking of single cells via positron emission tomography. Nat Biomed Eng. 2020;4:835-44
134. Gifani M, Eddins DJ, Kosuge H, Zhang Y, Paluri SLA, Larson T. et al. Ultra-selective carbon nanotubes for photoacoustic imaging of inflamed atherosclerotic plaques. Adv Funct Mater. 2021 31
135. Ronald JA, Kim BS, Gowrishankar G, Namavari M, Alam IS, D'Souza A. et al. A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant. Cancer Res. 2017;77:2893-902
136. Kim W, Le TM, Wei L, Poddar S, Bazzy J, Wang X. et al. [18F]CFA as a clinically translatable probe for PET imaging of deoxycytidine kinase activity. Proc Natl Acad Sci U S A. 2016;113:4027-32
137. Xin Y, Cai H. Improved Radiosynthesis and Biological Evaluations of L- and D-1-[(18)F]Fluoroethyl-Tryptophan for PET Imaging of IDO-Mediated Kynurenine Pathway of Tryptophan Metabolism. Mol Imaging Biol. 2017;19:589-98
138. De Silva RA, Kumar D, Lisok A, Chatterjee S, Wharram B, Venkateswara Rao K. et al. Peptide-Based (68)Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol Pharm. 2018
139. Gonzalez Trotter DE, Meng X, McQuade P, Rubins D, Klimas M, Zeng Z. et al. In vivo Imaging of the Programmed Death Ligand 1 by (18)F PET. J Nucl Med. 2017;58:1852-7
140. Natarajan A, Patel CB, Ramakrishnan S, Panesar PS, Long SR, Gambhir SS. A Novel Engineered Small Protein for Positron Emission Tomography Imaging of Human Programmed Death Ligand-1: Validation in Mouse Models and Human Cancer Tissues. Clin Cancer Res. 2019;25:1774-85
141. Lv G, Sun X, Qiu L, Sun Y, Li K, Liu Q. et al. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J Nucl Med. 2020;61:117-22
142. Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG. et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci U S A. 2015;112:E6506-14
143. Mayer AT, Natarajan A, Gordon SR, Maute RL, McCracken MN, Ring AM. et al. Practical Immuno-PET Radiotracer Design Considerations for Human Immune Checkpoint Imaging. J Nucl Med. 2017;58:538-46
144. Ehlerding EB, Lee HJ, Jiang D, Ferreira CA, Zahm CD, Huang P. et al. Antibody and fragment-based PET imaging of CTLA-4+ T-cells in humanized mouse models. Am J Cancer Res. 2019;9:53-63
145. Ehlerding EB, England CG, Majewski RL, Valdovinos HF, Jiang D, Liu G. et al. ImmunoPET Imaging of CTLA-4 Expression in Mouse Models of Non-small Cell Lung Cancer. Mol Pharm. 2017;14:1782-9
146. Wissler HL, Ehlerding EB, Lyu Z, Zhao Y, Zhang S, Eshraghi A. et al. Site-Specific Immuno-PET Tracer to Image PD-L1. Mol Pharm. 2019;16:2028-36
147. Chatterjee S, Lesniak WG, Nimmagadda S. Noninvasive Imaging of Immune Checkpoint Ligand PD-L1 in Tumors and Metastases for Guiding Immunotherapy. Mol Imaging. 2017;16:1536012117718459
148. Lesniak WG, Chatterjee S, Gabrielson M, Lisok A, Wharram B, Pomper MG. et al. PD-L1 Detection in Tumors Using [(64)Cu]Atezolizumab with PET. Bioconjug Chem. 2016;27:2103-10
149. Heskamp S, Hobo W, Molkenboer-Kuenen JD, Olive D, Oyen WJ, Dolstra H. et al. Noninvasive Imaging of Tumor PD-L1 Expression Using Radiolabeled Anti-PD-L1 Antibodies. Cancer Res. 2015;75:2928-36
150. Truillet C, Oh HLJ, Yeo SP, Lee CY, Huynh LT, Wei J. et al. Imaging PD-L1 Expression with ImmunoPET. Bioconjug Chem. 2018;29:96-103
151. Chatterjee S, Lesniak WG, Miller MS, Lisok A, Sikorska E, Wharram B. et al. Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem Biophys Res Commun. 2017;483:258-63
152. Kumar D, Lisok A, Dahmane E, McCoy M, Shelake S, Chatterjee S. et al. Peptide-based PET quantifies target engagement of PD-L1 therapeutics. J Clin Invest. 2019;129:616-30
153. Li D, Cheng S, Zou S, Zhu D, Zhu T, Wang P. et al. Immuno-PET Imaging of (89)Zr Labeled Anti-PD-L1 Domain Antibody. Mol Pharm. 2018;15:1674-81
154. Li D, Zou S, Cheng S, Song S, Wang P, Zhu X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol Pharm. 2019;16:3469-76
155. Stutvoet TS, van der Veen EL, Kol A, Antunes IF, de Vries EFJ, Hospers GAP. et al. Molecular Imaging of PD-L1 Expression and Dynamics with the Adnectin-Based PET Tracer (18)F-BMS-986192. J Nucl Med. 2020;61:1839-44
156. Jagoda EM, Vasalatiy O, Basuli F, Opina ACL, Williams MR, Wong K. et al. Immuno-PET Imaging of the Programmed Cell Death-1 Ligand (PD-L1) Using a Zirconium-89 Labeled Therapeutic Antibody, Avelumab. Mol Imaging. 2019;18:1536012119829986
157. Giesen D, Broer LN, Lub-de Hooge MN, Popova I, Howng B, Nguyen M. et al. Probody therapeutic design of 89Zr-CX-072 promotes accumulation in PD-L1 expressing tumors compared to normal murine lymphoid tissue. Clin Cancer Res. 2020: Epub Jan 17, pii: 1078-0432.CCR-19-3137. doi: 10.1158/078-0432.CCR-19-3137.
158. Natarajan A, Mayer AT, Reeves RE, Nagamine CM, Gambhir SS. Development of Novel ImmunoPET Tracers to Image Human PD-1 Checkpoint Expression on Tumor-Infiltrating Lymphocytes in a Humanized Mouse Model. Mol Imaging Biol. 2017;19:903-14
159. Natarajan A, Patel CB, Habte F, Gambhir SS. Dosimetry Prediction for Clinical Translation of (64)Cu-Pembrolizumab ImmunoPET Targeting Human PD-1 Expression. Sci Rep. 2018;8:633
160. Nagengast WB, de Korte MA, Oude Munnink TH, Timmer-Bosscha H, den Dunnen WF, Hollema H. et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51:761-7
161. van Es SC, Brouwers AH, Mahesh SVK, Leliveld-Kors AM, de Jong IJ, Lub-de Hooge MN. et al. (89)Zr-Bevacizumab PET: Potential Early Indicator of Everolimus Efficacy in Patients with Metastatic Renal Cell Carcinoma. J Nucl Med. 2017;58:905-10
162. Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J. et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6:53-8
163. Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R. et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017 9
164. Sellmyer MA, Richman SA, Lohith K, Hou C, Weng CC, Mach RH. et al. Imaging CAR T Cell Trafficking with eDHFR as a PET Reporter Gene. Mol Ther. 2020;28:42-51
Author contact
![]() Corresponding author: Neeta Pandit-Taskar, MD. Attending Physician, Molecular Imaging and Therapy Service, Department of Radiology, Memorial Sloan Kettering Cancer Center, Member, Memorial Hospital, New York, NY 10065. Professor, Department of Radiology, Weil Cornell Medical Center, New York, NY. Phone: (212) 639-3046 Pandit-norg
Corresponding author: Neeta Pandit-Taskar, MD. Attending Physician, Molecular Imaging and Therapy Service, Department of Radiology, Memorial Sloan Kettering Cancer Center, Member, Memorial Hospital, New York, NY 10065. Professor, Department of Radiology, Weil Cornell Medical Center, New York, NY. Phone: (212) 639-3046 Pandit-norg