- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2021; 5(4):378-390. doi:10.7150/ntno.54879 This issue Cite
Research Paper
Engineering Extracellular Vesicles to Target Pancreatic Tissue In Vivo
Hiroaki Komuro1,2,6*, Yuki Kawai-Harada2,6*, Shakhlo Aminova2,3, Nathaniel Pascual2,4, Anshu Malik2,6, Christopher H. Contag2,5,6, Masako Harada2,6 ![]()
1. Department of Cardiovascular Medicine, Tokyo Medical and Dental University, Tokyo, Japan.
2. Institute for Quantitative Health Science and Engineering (IQ), Michigan State University, Michigan, USA.
3. Lyman Briggs College, Michigan State University, Michigan, USA.
4. Department of Chemical Engineering and Material, Michigan State University, Michigan, USA.
5. Department of Microbiology and Molecular Genetics, Michigan State University, Michigan, USA.
6. Department of Biomedical Engineering, Michigan State University, Michigan, USA.
* These authors contributed equally to this work.
Received 2020-10-22; Accepted 2021-3-31; Published 2021-4-15
Abstract

Extracellular vesicles (EVs) are naturally released, cell-derived vesicles that mediate intracellular communication, in part, by transferring genetic information and, thus, have the potential to be modified for use as a therapeutic gene or drug delivery vehicle. Advances in EV engineering suggest that directed delivery can be accomplished via surface alterations. Here we assess enriched delivery of engineered EVs displaying an organ targeting peptide specific to the pancreas. We first characterized the size, morphology, and surface markers of engineered EVs that were decorated with a recombinant protein specific to pancreatic β-cells. This β-cell-specific recombinant protein consists of the peptide p88 fused to the EV-binding domain of lactadherin (C1C2). These engineered EVs, p88-EVs, specifically bound to pancreatic β-cells in culture and transferred encapsulated plasmid DNA (pDNA) as early as in 10 min suggesting that the internalization of peptide-bearing EVs is a rapid process. Biodistribution of p88-EVs administrated intravenously into mice showed an altered pattern of EV localization and improved DNA delivery to the pancreas relative to control EVs, as well as an accumulation of targeting EVs to the pancreas using luciferase activity as a readout. These findings demonstrate that systemic administration of engineered EVs can efficiently deliver their cargo as gene carriers to targeted organs in live animals.
Keywords: Extracellular Vesicles, Pancreatic β-cells, EVs engineering, Imaging, Targeted delivery.
Introduction
Over the last two decades, studies of extracellular vesicles (EVs) have shifted our understanding of their role in biology from early classifications of EVs as waste disposal machinery to a role in intracellular communication [1]. Cells secrete heterogeneous populations of lipid-bilayer membranous nano-sized particles such as exosomes, microvesicles (MVs) and apoptotic bodies whose composition may vary depending on the cell of origin, physiological and pathological condition of the cells or surrounding tissues [2]. While the sizes of these particles largely overlap with each other, the differences are consistent with the distinctive biogenesis of exosomes and MVs. Exosomes (40-150 nm in diameter) derive from the inward budding of endosomal multivesicular bodies (MVBs) and are released from the cell upon MVB fusion with the cell membrane. MVs (50-1,000 nm in diameter) are generally larger vesicles and are the product of direct budding from the plasma membrane [3, 4]. EVs are released from many different cell types into various body fluids, including milk, saliva, sweat and plasma, to mediate molecular transfer to other cell types in both physiological and pathological conditions [5-9].
A growing number of reports suggest small EVs (50 to 200 nm in diameter) facilitate the functional transfer of genetic material involved in diseases like diabetes, making EVs an appealing therapeutic gene delivery vehicle for immune therapy, vaccines, and regenerative medicine [9-20]. In fact, EV-mediated gene delivery circumvents the significant issues associated with synthetic nanoparticles, such as instability, immunogenicity, toxicity and biological barrier crossing [1, 21-24]. Naive EVs derived from stem cells or mesenchymal stem cells (MSCs) have ant-inflammatory, organ-protective and regeneration promoting properties, thus exert therapeutic effects in inflammation and organ injury [25-27]. Furthermore, several groups reported EV-mediated therapeutic delivery in vivo [28-31]. However, an EV engineering platform that can be readily modified for various cellular and tissue targets is essential for rapid development of EV-based therapeutics with clinical applications. Such a platform should be designed to overcome technical limitations, including a lack of controlled generation of EVs, inefficient loading, and inadequate tissue specificity [32-34]. EV surface proteins contribute to EVs' natural tropism, hence, the modification of these proteins to include surface adhesion molecules and ligands, may direct specific binding to desired cells, tissues or organs [22, 30, 35-38]. There are several methods proposed for ligand display on EV surface. One such method fuses targeting moieties to known EV membrane proteins such as Lamp2b and CD63 [31, 39-41]. Alternatively, the C1C2 domain of the phosphatidylserine (PS) binding protein milk fat globule-EFG factor 8 (MFG-E8), also known as lactadherin, enables versatile EV surface display since it can be used to decorate the EV surface when expressed from producer cells, or when added to purified EVs [42]. Several groups have shown that lactadherin C1C2 can modify exosome surfaces and target fusion proteins, such as anti-EGFR nanobody, carcinoembryonic antigen and HER2 antibody, to EVs [22, 38, 43-45]. It has been demonstrated that producer cells, transfected with an expression plasmid, package pDNA into EVs, and if that plasmid encodes a surface protein that is expressed on EVs then the EVs will contain the DNA representing the surface expressed protein [20, 46, 47]. EVs from cells containing such pDNAs would then have a feature of bacteriophages that make phage display screens possible. Towards developing an in vivo EV display screening protocol, we evaluated tissue targeting and EV packaging in this study.
Here, we used C1C2 to display p88, a peptide with known affinity for the human and mouse membrane small ion transport regulator (FXYD2)γa, to generate pancreas targeting EVs. A contrast agent (CA) consisting of p88 with a chelator and GdCl3 showed a potential β-cell specificity as an imaging agent for the MRI (magnetic resonance imaging), suggesting p88 as a potential β-cell targeting peptide [48, 49]. We demonstrate that β-cell targeting EVs can enrich pDNA delivery to the pancreas in living mice. The pancreas is an inaccessible organ for both surgical and drug treatment due to its anatomical position, and potential experimental drugs are not clinically applicable due to the complications derived from the toxicity or inefficient delivery [50-52]. This study proposes a unique technique for EV-mediated targeted delivery to pancreatic tissue and supports the development of customizable EV-based targeted delivery vehicles for nucleic acid-based therapeutics.
Materials and Methods
EV-peptide display plasmid construction
Seamless Ligation Cloning Extract (SLiCE) mediates in vitro DNA assembly through a RecA-independent recombination mechanism between DNA fragments with short homologous ends [53, 54]. The SLiCE reagent, a bacterial lysate, was prepared from E.coli DH5α strain and used for all the cloning in this work following a previously described procedure [53, 54]. Briefly, EV-display constructs were created by SLiCE assembly of PCR fragments into pcDNA6.0 V5/His (Invitrogen) digested with NheI and AgeI. The signal peptide and lactadherin C1C2 domain were amplified from psd44-Lactadherin46 (a gift from Agnese Mariotti Addgene, plasmid # 46830) using the primer sets (Table S1) which included overhangs. The coding regions of mCherry fluorescent protein and the gaussia-luciferase (gLuc) bioluminescent protein were amplified from pLM-CMV-R-Cre [55] (Addgene plasmid #27546) and pcDNA3.1(+)-GLuc (a gift from Contag lab), respectively. The PCR fragment was amplified from the pcDNA-mCherry-C1C2 plasmid and assembled with the synthetic double-stranded oligonucleotide consisting of pep1 coding sequence and the (GGGGS)3 linker sequence [56] (pep1-3xG4S-C1C2) to generate pep1-EV display construct. Similarly, a fragment was amplified from the pep1-EV display construct and assembled from synthetic oligonucleotides consisting of p88 coding sequence with homologous ends (P885-3-1 and P885-3-2) to create p88-EV display construct. pcDNA backbone for both pep1 and p88 was further down-sized by removing unnecessary sequences including the mammalian selectable marker (Blasticidin) and the phage origin of replication through the single-piece SLiCE reaction of the PCR fragments. This resulted in pcS-p88-C1C2 and pcS-pep1-C1C2. Another fragment was amplified from the pcS-p88-C1C2 using a primer set (HA-3xG4S-F and HA-R) and assembled to generate Non-Peptide display construct, pcS-NP-C1C2. The primers and oligos are listed in Table S1. Each construct was purified using the QIAprep Miniprep kit (QIAGEN) and sequenced for validation. The Plasmid Plus Midi Kit (QIAGEN) was used for large scale plasmid preparation.
Cell culture and treatment
The following cell lines, obtained from American Type Culture Collection (ATCC), were tested for mycoplasma: 293T (Human Embryonic Kidney cell line), NIT-1 (Mouse pancreatic β-cell line) and 4T1 (murine mammary carcinoma cell line). The cells were cultured in high-glucose DMEM (Gibco) for 293T or RPMI-1640 (Lonza) for 4T1 media supplemented with 100U/mL penicillin, 100µg/mL streptomycin and 10% (v/v) fetal bovine serum (FBS, Gibco). FBS was ultracentrifuged in PET Thin-Walled ultracentrifuge tubes (Thermo Scientific 75000471) at 100,000g with a Sorvall WX+ Ultracentrifuge equipped with an AH-629 rotor (k factor = 242.0) for 18 h at 4 °C to remove the bovine EVs and create EV-depleted FBS for use in the culture media for preparation of engineered EVs. NIT-1 cells were cultured in F12 (Mediatech Inc.) media supplemented with 100U/mL penicillin, 100 µg/mL streptomycin and 10% (v/v) fetal bovine serum (Gibco). All cells were maintained in a humidified incubator with 5% CO2 at 37 °C. For EV production, EV-display constructs were either transfected alone or along with an imaging EV-display plasmid into 293T cells. In-house PEI (polyethylenimine, Sigma 408727) transfection reagent was used, which works similarly to commercially available polymer- or liposome-mediated in vitro transfection reagents [57]. Cells were seeded at 2x106 in a 10cm cell culture dish for 24 h in regular culture media and transfected with 10 µg total DNA suspended with PEI in non-supplemented DMEM. To prepare the DNA-PEI transfection mixture, 10 µg DNA/100 mm dish was added to PEI in a ratio of 1:2.5 (DNA:PEI) in non-supplemented DMEM, pulse-vortexed for 30 sec, and incubated at room-temperature for 10 min. Following 24 h incubation, cells were washed twice with PBS, and the culture media was replaced with DMEM supplemented with EV-depleted-FBS for another 24 h incubation for EV production. For naïve EV production, cells were cultured with DMEM supplemented with EV-depleted FBS without transfection for 24 h. mCherry labeled and p88-mCherry co-labelled EVs were prepared from 293T cells transfected with the mCherry-EV display construct (pcDNA-mCherry-C1C2) and co-transfected with mCherry-EV display and p88-EV display constructs. EVs labeled with gaussia-Luciferase (gLuc), co-labelled with p88-gLuc, co-labelled with PEP1-gLuc and co-labeled with non-peptide (NP)-gLuc were prepared by transfecting 293T with plasmid pcD-gLuc-C1C2 and co-transfection with pcS-p88-C1C2 [58].
EVs isolation
The cells were grown in DMEM media supplemented with EV-depleted FBS for 24 h, and the media from the plates was collected. For each batch, EVs were purified from 36 mL of conditioned media by differential centrifugation. The media was centrifuged at 400g for 10 min and then 600g for 30 min to remove the cell and cell debris. In order to remove the contaminating apoptotic bodies, the media was centrifuged at 2000g for 30 min. The supernatant was then ultracentrifuged in PET Thin-Walled ultracentrifuge tubes (Thermo Scientific 75000471) at 100,000g with a Sorvall WX+ Ultracentrifuge equipped with an AH-629 rotor (k factor = 242.0) for 90 min at 4 °C to pellet the EVs [59]. The pellet containing EVs was resuspended in 100 µL PBS or PBS with 1% trehalose [60] except the gLuc labeled EVs, which were resuspended in DPBS (PBS with calcium and magnesium; Gibco 14190136).
Nanoparticle Tracking Analysis (NTA)
The particle size and concentration were measured using a ZetaView® (Particle Metrix) Nanoparticle Tracking Analyzer following the manufacturer's instruction. EVs were diluted in PBS between 100- and 1000-fold to obtain a concentration within the recommended measurement range (0.5X105 to 1010 cm-3).
Western Blotting
Cells (transfected and non-transfected) were lysed by MRIPA lysis buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.25% sodium deoxycholate, 50 mM Tris, pH 7.4), and the supernatant was used as cell lysates. Protein concentration was measured by Micro BCA Protein Assay kit (G Biosciences) using BSA as a standard. 50 µg of the protein and 1x109 EVs were denatured at 70°C for 10 min in NuPAGE LDS Sample Buffer (Thermo Fisher Scientific), separated on a 12% SDS PAGE, and transferred to a nitrocellulose membrane. The membrane with the blotted proteins was blocked with blocking buffer containing 5% milk in Tris-buffered saline (TBS) for 2 h and then incubated with a primary antibody at 4°C overnight. Following three washes with TBS with 0.1% Tween 20 (TBST), the membrane was incubated with secondary horseradish peroxidase-conjugated secondary antibody for 1.5h at room temperature. The membrane was again washed thrice with TBST and the protein bands were visualized by treating with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Scientific) and the image was captured by ChemiDoc Imaging System (BioRad). The following primary antibodies were used: anti-HA (Sigma Aldrich, H3663), anti-β-actin (Sigma Aldrich, A5441), anti-CD63 (Thermo Fisher, 10628D), anti-TSG101 (Abcam, ab125011), and anti-calnexin (Abcam, ab22595). The following secondary antibodies were purchased from Invitrogen: Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP (A16078) and Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP (A16110).
Immuno-Transmission electron microscopy (Immuno-TEM)
Carbon film coated 200 mesh copper EM grids were soaked in 50 µL EVs (1x107 Naïve, p88 and pep1 EVs in PBS) for 30 min for the adsorption of EVs on the grid. EVs on the grids were fixed by treating with 50 µL of 2% Paraformaldehyde (PFA) for 5 min and then rinsed thrice with 100 μL PBS. To quench free aldehyde groups, the grids were treated with 50 μL of 0.05 M glycine for 10 min. The surface of the grids was blocked with a drop of blocking buffer (PBS containing 1% BSA) for 30 min. After blocking, the grids were incubated with 50-100 μL anti-HA (Sigma-Aldrich H3663) or anti-CD63 (Thermo Fisher 10628D) antibody (1:100 in PBS containing 0.1% BSA) for 1 h. The grids were washed five times with 50 μL PBS containing 0.1% BSA for 10 min each. For secondary antibody treatment, the grids were incubated in a drop of Goat-anti-Mouse IgG coupled with 10 nm gold nanoparticles (Electron Microscopy Sciences, 25512) diluted at 1:100 in PBS containing 0.1% BSA for 1 h. The grids were washed five times with 50 μL PBS containing 0.1% BSA for 10 min each and then with two separate drops of (50 μL) distilled water. EVs were negatively stained with 2% uranyl acetate and then rinsed with PBS. The grids were then air dried for 24-48 h and images were captured by Transmission electron microscope (JEOL 1400) at 80 kV.
Confocal Microscopy
2.5x106 naïve, mCherry labeled, and p88-mcherry co-labeled EVs were loaded on a coverslip at three different locations. Dried and mounted EVs were then analyzed under a fluorescence microscope (Nikon Eclipse Ts2R) at 60X magnification. NIT-1 cells were cultured until passage 18 to ensure positive labeling with insulin [61]. For EV binding studies, cocultured NIT-1 cells and 4T1 (3X104) cells in a 4-well chamber slide (Nunc Lab-Tek) were treated with 1x107 mCherry-EVs or mCherry co-labeled p88-EVs for 1 h at room temperature. Cells were rinsed with 1X PBS containing 0.1% Tween 20 to remove unbound excess EVs. To fix the cells, the slide was treated with 250 µL 4% PFA at room temperature for 10 min. The cells were washed thrice with ice-cold PBS, and treated with blocking buffer (1% BSA, 22.52 mg/mL glycine in PBST (PBS+ 0.1% Tween 20)) for 30 min. Then they were incubated in a humidified chamber with 250µL anti-insulin antibody (Guinea pig polyclonal Insulin antibody, Abcam ab7842; 1:100 diluted 1% BSA in PBST) overnight at 4°C. To remove unbound antibodies, the cells was washed thrice with PBS for 5 min each. Then they were treated with 250 µL solution of secondary antibody (Goat Anti-Guinea pig IgG H&L (FITC), Abcam ab6904; 1:1000 diluted in 1% BSA in PBST) for 1 h at room temperature in dark. After removing secondary antibody solution, the cells were washed thrice with PBS for 5 min each in dark. For counter staining, the cells were treated with 250 µL of 0.5 µg/mL solution of DAPI for 1 min. The cells were again rinsed with 1X PBS, and then coverslip was mounted on the cells after applying mounting medium (Life Technologies, P36930). The slide was stored at 4°C in the dark for further analysis of the cells using a confocal fluorescence microscope (Nikon A1Rsi).
In vitro Bioluminescent assay
In this assay, naïve EVs, NP-gLuc EVs and p88-gLuc EVs were placed in wells of a 96 well plate (UV-Star® Microplate, 96 well, COC, F-Bottom (Chimney Well), uClear®, Clear; Greiner Bio-one) in triplicate. 95 µL of DPBS was added to each well and then treated with 50 µL 1 µg/mL Coelenterazine-H (CTZ; Regis Technologies). The luminescence was recorded using an in vivo imaging system (IVIS; Spectrum Perkin Elmer). For control 5µL of DPBS was used and treated in the same manner.
In vitro Bioluminescent assay
The EV-binding assay was performed using bioluminescent imaging (BLI). NIT-1 mouse β-cell mouse pancreatic β-cells and 4T1 mouse mammary carcinoma cells were seeded at 1.0x104 cells/96-well plates 24 h prior to EV treatment. The cells were fixed with 4% PFA at room temperature for 10 min and washed three times with PBS. The cells were treated with 1.0x107 NP-gLuc-EVs or p88-gLuc-EVs in 100 µL media for 0, 30 and 60 min at 37 °C. Following the two PBS washes to remove unbound EVs, CTZ (1 µg/mL in PBS) was added to the wells and imaged by IVIS. Total photon flux (photons/sec) was quantified using Living Image 4.7.2 software (IVIS, PerkinElmer). Values are presented as the means ± SD (n = 3).
Animals
In this study, female Balb/cJ mice (6 weeks old) were used for animal experiments. Animals were purchased from Jackson Laboratories and housed in the University Laboratory Animal Resources Facility at Michigan State University. All the experimental procedures for the animal study were performed with the approval of the Institutional Animal Care and Use Committee of Michigan State University.
Ex vivo imaging of mice organs
Anesthetized mice received intravenously injection of 1.0x1010 NP-gLuc EVs, p88-gLuc EVs or PBS (n=2). Following 30 min circulation, the mice were sacrificed and the following visceral organs were dissected and placed on a transparent sheet: heart, lungs, liver, kidneys, pancreas and spleen. Ex vivo images of BLI were taken following re-application of 200 µL CTZ (10 µg) to the resected organs by IVIS. The images were quantified using Living Image 4.5 software (IVIS, PerkinElmer).
Plasmid DNA Recovery from Animal Tissue
Approximately 1.0x109 NP-EVs (n=2) or p88-EVs (n=3) in 100 µL PBS were intravenously injected into mice. Following 1 h of EVs administration, the mice were sacrificed, and the visceral organs (heart, lung, liver, kidney, pancreas and spleen) were dissected and homogenized using Triple-Pure High Impact 2.8mm Steel Beads (Benchmark Scientific) and BeadBug 6 Microtube Homogenizer (Benchmark Scientific). The plasmid DNA was isolated from the organ homogenates using QIAprep Spin Miniprep Kit following a modified protocol for plasmid isolation from mammalian cells that would exclude chromosomal DNA [62]. The copy number of the plasmids was assessed by qPCR-based TaqMan assay.
DNase I Treatment
The 2 µL of Naïve EVs mixed with 107 copies of pDNA or p88-EVs were incubated at room temperature for 15 min with 1 U of DNase I (Zymo Research) and DNA Digestion Buffer. The plasmid DNA was isolated from the EVs using Qiamp Miniprep kits and quantified by qPCR.
Quantitative Real-time Polymerase Chain Reaction (qPCR)
Evaluation of Plasmid DNA associated with EVs
qPCR was performed using Taq DNA polymerase (Fisher BioReagents). Each reaction contains 200 µM dNTP, 500 nM each of forward/reverse primer, 400 nM probe (Table S1), 0.5 U Taq DNA polymerase, 1x Assay buffer A and 1 µL sample DNA or isolated EV in a total reaction volume of 10 µL using CFX96 Touch Real-Time PCR Detection System (BIO-RAD). The PCR amplification cycle was as follows: 95°C for 2 min; 40 cycles of 95°C for 20 seconds, 65°C for 30 seconds. The pDNA copy number were determined by absolute quantification using the standard curve method, and the copy number of EV encapsulated pDNA per vesicles was calculated based on NTA and qPCR results.
Small DNA Recovered from each Organ
qPCR was performed using PrimeTime® Gene Expression Master Mix (Integrated DNA Technologies, Inc.). Each reaction contains, 500 nM each of forward/reverse primers, 200 nM probe (Table S1), and 1 µL sample DNA in a total reaction volume of 10 µL using CFX96 Touch Real-Time PCR Detection System (BIO-RAD). The PCR amplification cycle was as follows: 95°C for 2 min; 40 cycles of 95°C for 15 seconds, 65°C for 30 seconds. The recovered pDNA from each organ was normalized by recovered Mitochondria DNA.
Mitochondria DNA detection
qPCR was performed using Phusion® High-Fidelity DNA Polymerase (New England Biolabs, inc.). Each reaction contains, 0.2 U Phusion® High-Fidelity DNA Polymerase, 1x Phusion HF buffer, 200 uM dNTP, 1/20,000 diluted SYBR™ Green I (Invitorogen), 500 nM each of forward/reverse primer (Table S1), and 1 µL sample DNA in a total reaction volume of 10 µL using CFX96 Touch Real-Time PCR Detection System (BIO-RAD). The PCR amplification cycle was as follows: 98°C for 2 min; 40 cycles of 98°C for 10 seconds, 60°C for 20 seconds, 72°C for 20 seconds. The size of PCR product was analyzed by agarose gel electrophoresis.
Results
Design, Generation and characterization of engineered EVs displaying pancreatic β-cell targeting peptide
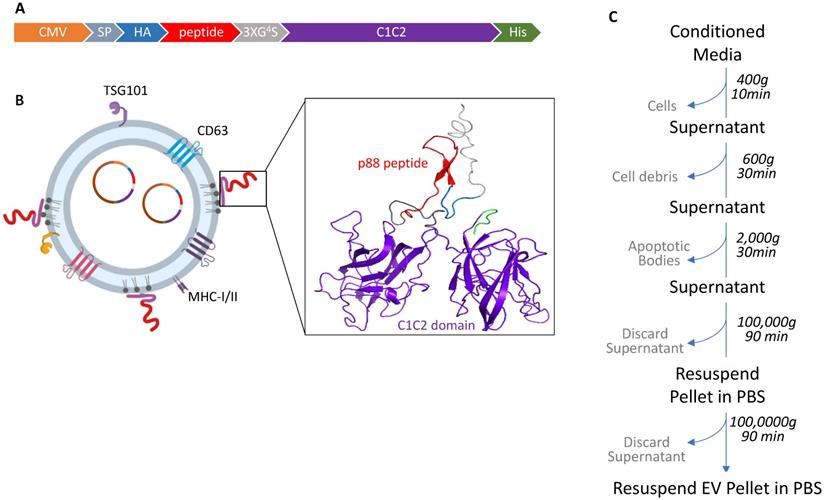
We investigated whether EVs can display β-cell-targeting peptides on their surface to improve specificity and enhance EV-mediated cargo delivery to the target cells after systemic administration in living animals. The p88 peptide, known to bind (FXYD2)γa on pancreatic β-cells, was used as a targeting ligand for pancreatic β-cells [48]. The pep1 peptide raised against p16-overexpressing cancer cells served as a control peptide with a negligible affinity for pancreatic cells in our system [63]. In order to label EVs with these peptides, we fused peptide sequences followed by the (GGGGS)3 linker with the C1C2 domain of lactadherin (Fig. 1A and 1B). Following transfection, the EV fraction was collected from the culture media by ultracentrifugation, as described in Figure 1C.
Design and schematic presentation of a C1C2-peptide fusion protein (A) Peptide-C1C2 fusion protein expression driven by a CMV promoter in pcDNA6.0 derived pcS vector. The recombinant protein comprising a lactadherin signal peptide (SP), Hemagglutinin tag (HA), peptide sequence, (GGGGS)3 Linker, EV anchor region of lactadherin (C1C2), and polyhistidine tag (HA). (B) Depiction of EV with engineered EV displaying peptide on its surface and encapsulated pDNA, and predicted protein structure of peptide-hC1C2 chimeric protein. (C) Schematic flow of EV isolation process.
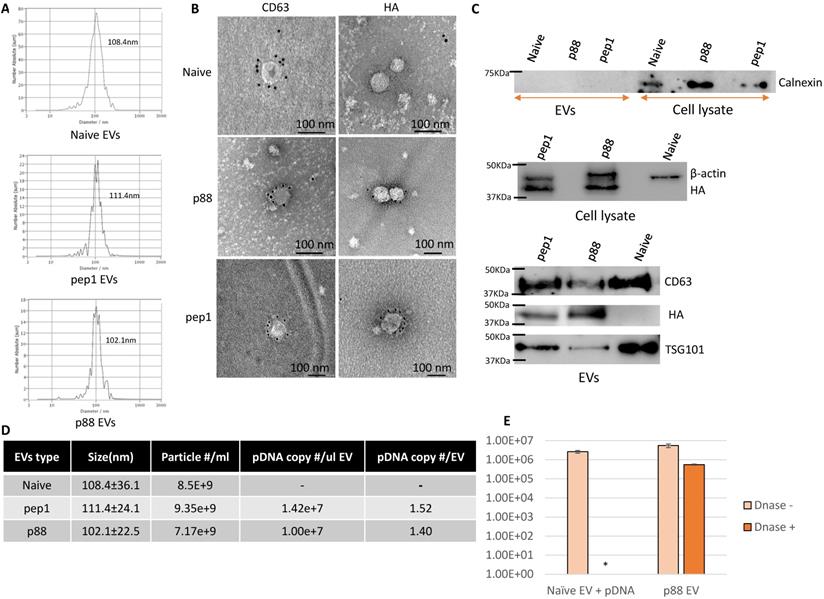
In addition to being devoid of disease-related molecules, the engineered EVs generated from 293T cells projected minimal toxicity and immunogenicity effects in mice [64, 65]. These EVs were characterized following MISEV (Minimal Information for Studies of Extracellular Vesicles 2018) guidelines [66]. The particle size of 293T-derived EVs peaks around 100 nm at the concentration ranges from 1-9 x109 particles/mL (Fig. 2A). The presence of peptide-C1C2 did not affect EV size or morphology (Fig. 2A and 2B). Immuno-TEM images demonstrated typical EV morphology with the presence of EV marker (CD63) and peptide (HA) on the engineered EVs (Fig. 2B and S1). As shown in Figure 2C, both p88-C1C2 and pep1-C1C2 fusion proteins were successfully expressed in these cells and on EVs, which appeared as bands proximal to their calculated molecular weights of 43.9 kDa and 42.0 kDa, respectively. Furthermore, all the EV types were positive for EV markers CD63 and TSG101, whereas cell-specific protein calnexin was not detected, further confirming the purity of the EVs without cell contamination. qPCR analysis using a probe specific to the encapsulated pDNA in combination with nanoparticle tracking analysis determined the quantity of pDNA in these vesicles. Notably, reducing pDNA size by removing non-essential sequences improved transfection efficiency and subsequent pDNA packaging efficacy (data not shown). Figure 2D shows representative data of engineered EVs analyzed by NTA particle count and qPCR data generated by a primer and probe set over plasmid specific regions. There were copy number variabilities in each batch of EVs, but the results consistently showed 0.3-2 pDNA copies per EV. Further, we examined the number of plasmids encapsulated inside of the engineered vesicles by EV-DNaseI treatment. As shown in Figure 2E, it removed approximately 90% of the plasmids, indicating that 10% of the total plasmid were encapsulated. Thus, the engineered EVs incorporate the majority of the DNA on their surface.
Successful isolation and characterization of engineered EVs displaying peptides of interest. (A) Representative size distribution of the naïve, pep1- and p88-display EVs determined by Nanoparticle Tracking Analysis. (B) Transmission electron microscopy images of naïve, pep1-, and p88-EVs showing gold labeled HA and CD63 surface markers. (C) Western blot analysis of engineered EVs (p88 fusion peptide-44Kda; pep1 fusion peptide-42KDa) for the presence of EV biomarkers CD63(30-60KDa) and TSG101(44KDa), and peptide HA-tag. Additionally, analysis of cell lysate and engineered EVs for cellular biomarkers Calnexin (67KDa) and β-actin (42KDa) (D) Summary of particle number and pDNA copy numbers determined by NTA and qPCR of pep1- and p88-display EVs. (E) pDNA quantification before and after DNase I treatment of EVs. Naïve EVs mixed with pDNA and p88 EVs were treated with DNase I. pDNA were quantified by qPCR following pDNA isolation. *pDNA undetected
Binding of targeting EVs to NIT-1 pancreatic β-cells in vitro
GLuc labeled targeting (p88) and non-targeting (NP) (Fig. S2) were evaluated on the cultured cells using bioluminescence imaging (BLI) to examine the binding capacity of β-cell targeting EVs. NIT-1 mouse pancreatic β-cells and 4T1 mouse mammary carcinoma cells were treated with p88- or NP-EVs following PFA fixation. NIT-1 cells showed stronger bioluminescence after 30 min incubation with p88-gLuc-EVs, compared with NP-gLuc-EV-treated cells, demonstrating a higher binding capacity of p88-EVs towards β-cells compared to Non-peptide-EVs (Fig. 3A). There were very few signals detected for non-β (4T1) cells treated neither with p88- nor NP-EVs. To further evaluate the binding capacity towards β-cells, binding was observed using NIT-1 cells co-cultured with 4T1 cells.
Specific binding of targeting EVs to pancreatic β-cell line in vitro. (A) NIT-1 cells and 4T1 cells received non-tarting (NP)- or β-cell-targeting (p88)-EV treatment after PFA fixation. Representative image of EV (gLuc) binding to NIT-1 or 4T1 cells. The total photon flu (p/s) from EVs bound to the cells was quantified using IVS. The value represents the means ± SD (n=3) in the graph. (B) NIT-1 and 4T1 cells were co-cultured and treated either with mCherry-EVs (upper row), or p88-mCherry-EVs (lower row) for 1 hr. The cells were fixed, and the binding was assessed by confocal imaging of EVs (red), anti-insulin antibody (FITC-conjugated) and nuclear staining with DAPI. microscopic images of cocultured NIT1 (FITC insulin labeled; Green), 4T1 cells treated with p88-mCherry-EVs, and mCherry-EVs (red). DAPI stain (blue) Scale bars, 20 μm.
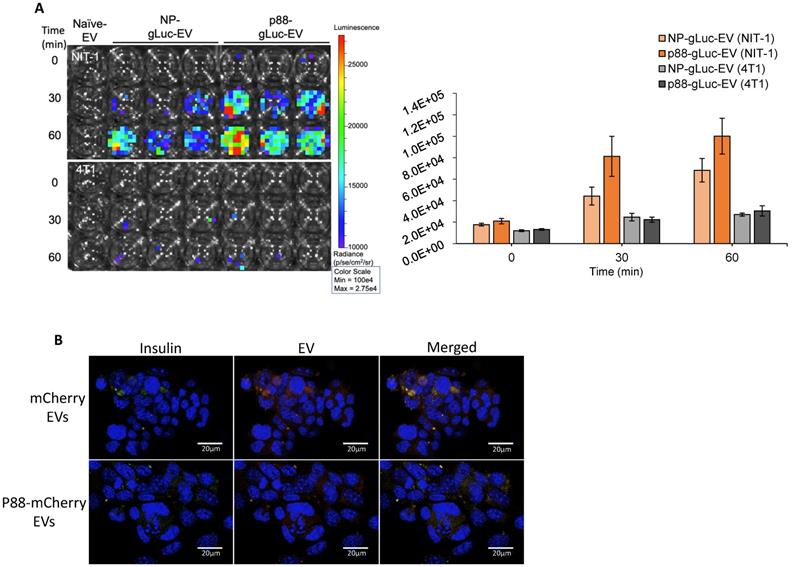
EVs were labelled with mCherry (mCherry-EVs) or co-labelled with p88 and mCherry (p88-mCherry EVs) (Fig S3) prior to the treatment. After 1 h of incubation, the p88-mCherry-EVs (red) mainly localized to the insulin-secreting β-cells (green). In contrast, the EVs without p88 (red) bound non-specifically regardless of cell type (Fig. 3B), indicating peptide mediated EV binding to the β-cells. Time-course experiments using NIT-1 cells to determine pDNA uptake using p88-EV and pep1-EV showed that the rate of pDNA uptake was not distinguishable between two EV types (Fig. S4A). The further analysis using the non-peptide (NP-EVs) version of the construct showed similar trends (Fig. S4B), suggesting the limitation of the DNA uptake assay with the static and restricted flow of the tissue-culture cellular environment. The uptake usually starts as early as 10 min for targeting EVs and saturate at 1 h time point for both non-targeting and targeting EVs.
Biodistribution of pancreas targeting by engineered EVs
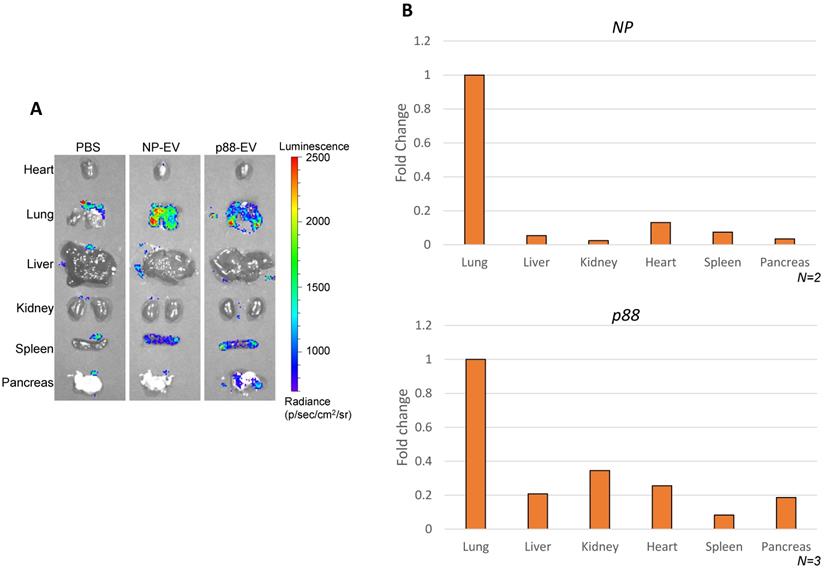
Biodistribution of targeting (p88-gLuc) and non-targeting (NP-gLuc) EVs in live mice were evaluated by ex vivo imaging. A high background signal of the substrate was captured in the control mice organs treated with PBS (Fig. 4A), which was indistinguishable from the signals received from mice treated with engineered EVs as previously reported [67]. The signals from the internal organs of the respective mice (Fig. 4A and S5A) verified the circulation of EVs in the body. The signals from the non-targeting EVs were primarily detected in the lung and spleen. The bioluminescence signals representing targeted EVs were predominantly observed from the lung, spleen and pancreas, implying the affinity-mediated navigation of EVs to the pancreas following the typical circulation pattern through the lungs and heart. The background luminescence of the substrate was spotted only in the lungs of control mice, and faded upon dilution in blood circulation by heart, as no signal was detected in other organs. The experiment verified directed steering of targeting EVs by the displayed peptide to the pancreas through systemic blood circulation.
Altered EV biodistribution by peptide-display, and pancreas enriched pDNA delivery by β-cell-targeting EVs. (A) Representative images of the organs from the Balb/cJ mice received intravenous injections of PBS, NP-gLuc- or p88-gLuc-EVs. (B) Balb/cJ mice received intravenous injection of NP- or p88-labeled EVs. The organs were removed from the mice post-mortem and homogenized for pDNA isolation. qPCR assay was used to determine the copy number from heart, lungs, liver, kidneys, pancreas and spleen. The amounts of recovered pDNA were normalized by mitochondrial DNA. The fold change values represent the average fold change of samples (N).
EV-mediated targeted delivery of pDNA in vivo
The ability of p88-EVs to selectively deliver pDNA in vivo was assessed by recovering and quantifying pDNA from pancreatic homogenates of mice 1 h post-administration with targeting EVs. pDNA recovery from organs were confirmed by the mitochondrial DNA (mtDNA) qPCR (Table S2). Strikingly, the pancreas of mice that received pancreas targeting p88-EVs exhibited the accumulation of p88-coding pDNA in contrast to the spleen, as shown in Figure 4B. We further demonstrated that assayed pDNA are indeed plasmid and not fragmented DNA pieces by qPCR analysis using the primer sets over 2 other regions of the plasmid (ampicillin gene and C1C2-coding region). All of the qPCR assays used in this work uses primer sets amplifying the peptide-coding region of the EV-display plasmid (Fig. S5B).
Discussion
There is significant research interest in developing EV therapeutics that harness the innate ability of EVs to mediate the delivery of a diverse selection of cargos [9, 10, 12, 23-25, 32-34, 68]. Unlike synthetic nanocarriers, EVs feature more robust stability in vivo since they do not provoke strong immunogenic responses or toxic side effects [1]. Furthermore, EVs can be engineered with relative ease by fusing targeting moieties to known EV surface proteins like Lamp2b, tetraspanins (CD63, CD81, CD53, CD37, and CD82), and Lactadherin [37, 39-41, 69]. In this work, we demonstrated targeted delivery of pDNA to the pancreas using engineered EVs that display a β-cell specific peptide in vivo. We also analyzed the EV targeting by in vivo bioluminescence imaging and exogenous EV-mediated delivery by quantifying the pDNA copy number within engineered EVs and resected organs.
Through the surface display of the p88 peptide, the engineered EVs attained affinity towards the ion transport regulator (FXYD2)γa displayed on pancreatic islets [48, 49, 70]. The p88 peptide, in combination with a Gadolinium-based contrast agent, was previously developed for non-invasive in vivo imaging and quantification of β-cells in MRI studies [48, 49]. We showed EVs engineered to display p88 (p88-EVs) exhibited higher binding capacity to β-cells in co-cultured cells in vitro (Fig. 3B). The in vitro DNA uptake study showed high variabilities indicated by the large error bars, suggesting the limitation of the in vitro assay (Fig. S4A, B). This could be due to the restricted cultured environment cause uptake and release of exogenous pDNA in the simultaneous manner. Thus, the in vitro assay does not represent the biological response in vivo and is not conclusive.
Molecular display using lactadherin C1C2 fusion proteins was used to improve EV targeting in several studies [22, 42, 71-73]. While the exact mechanism of C1C2 localization to EV membranes still remains unclear, the use of C1C2 fusion proteins, when bound to PS on EV membrane surfaces, provides additional benefits such as inhibiting the recognition of PS by coagulation factors and macrophages [43, 44]. Thus, purified C1C2-fusion proteins can be reconstituted with isolated EVs to engineer the surface of EVs in a plug-and-play manner [42, 73].
Pancreatic β cells are relatively inaccessible as a drug target due to their deep anatomical location and structure on the islet. This limits diagnostic and therapeutic options. Previous attempts to target the pancreas, for example apratoxin A, showed high antiproliferative capabilities, though it caused notable in vivo toxicity [50]. We show that in vivo systemic administration of β-cell targeting EV harnessed targeting delivery of cargo pDNA to the pancreatic cells (Fig. 4B). Importantly, to our knowledge, this is the first study to show the recovery and quantification of exogenously introduced pDNA cargo to the pancreas, whereas previous studies demonstrated the physiological changes without clearly determining the type of molecules (DNA/RNA/protein) delivered to their target cells [30, 39, 74, 75]. The efficient DNA recovery was confirmed by co-isolation of high-quantity mitochondrial DNA (Table S2).
While (FXYD2)γa, which was used in this study, has been identified as a specific biomarker to pancreatic β-cells, its expression is not limited to the pancreas. This ion transport regulator has been found in the liver, kidney, salivary gland and gallbladder of humans, in addition to the pneumocytes of macaques [48, 49, 70, 76]. This could possibly explain pDNA delivery by p88-EVs to non-target organs (Fig. S4), but the total number of copies of pDNA delivered to the pancreas remains significantly higher compared other organs (Fig. 4C). Although the p88-pDNA recovery from the pancreas was consistently high from tested animals, the pDNA copy number has variabilities between animals and among organs, and at 2000 copies per pancreas, efficiency could be improved. The variability could be due to circulation time, sample processing and variabilities between EVs quality from the storage durations and conditions, which require improvements.
The confocal microscopic imaging of the EVs in co-cultured cells clearly demonstrated that p88-EVs binds to pancreatic β-cells in culture within 1 h (Fig. 3B). EVs were labeled with mCherry using endogenous mCherry-C1C2 pDNA transfection method, and fluorescence was verified prior to treatment (Fig. S3).
In vivo EV imaging was also used to demonstrate targeting, but their nanosized and labeling limitations complicate EV visualization. For example, fluorescent lipophilic dyes (PKH67, PKH26, R18, Dil, and DiD) stain the EV membrane, but only allow for imaging after harvesting the organ due to its low sensitivity [36, 77-79]. Furthermore, lipophilic dyes persist in tissues or circulation even after the degradation or dissociation of EV's; so, in the case of using lipophilic dyes, EVs must be differentiated from the dye itself and additional background signals [36, 69, 80, 81]. Radiolabeling of EVs, using 99mTc, 131I, and 111In-oxine, are promising in non-invasive biodistribution studies, offering both quantitative and qualitative data [80, 82, 83]. However, there are still some limitations to overcome, such as short half-life and inefficient EV recovery post labeling. In this study, the luciferase from Gaussia princeps, a reporter protein that emits bioluminescence in the presence of its substrate coelenterazine, was used; this was similar to the approach described by Takahashi et al [58]. In line with their findings of EV monitoring in live mice, we observed a similar pattern of diffuse signals over the ventral side of mice and absence of any localized signal. However, our bioluminescent signals did not extend up to 30 mins post substrate injection. Although, we used similar bolus i.v. injection of the substrate, we only detected signals until 10-15 mins post substrate injection. In addition to potential nonspecific interference from vascular epithelial cells or blood cells, the presence of FXYD2 receptors in other tissues such as salivary and mammary glands, dorsal root ganglia, and kidney could cause the observed signal scattering.
The importance of EV-mediated DNA delivery lies in the potential of directed transfer of therapeutic DNA in vivo. The methods used in our work combined simple well-established methods to show targeted pDNA delivery, including qPCR DNA quantification from EVs and the harvested organ. Alternatively, next-generation sequencing to identify and quantify all RNA biotypes or shotgun proteomics could have been used to provide broad-scale data analysis [79, 84]. Targeting is key to effective delivery of therapeutics allowing precise localization to diseased tissues and thus eliminating side effects derived from off-target effects of large drug dosage.
There are still inefficient and variable loading and DNA into EVs and recovery among sets of experiments, but our data holds promise for future customized therapeutics. Precise targeting may allow for guided-tissue regeneration by delivering genes to stem cells in targeted tissues. Similarly, this approach could deliver genes to create producer cells in target tissues to generate bystander effects which can influence groups of surrounding cells as was reported by Kanada et al [28]. As mentioned, efficient gene packaging and generation are limitations yet to be overcome for effective EV-mediated DNA delivery. EV generator cells with engineered surface ligands cloned into the genome may allow efficient EV production at scale. Electrical stimulation of cells (live cellular nanoporation) to efficiently package mRNA to EVs and to increase EV production would increase EV numbers from producer cells [85]. The combinations of such cutting-edge technologies will shed lights to targeted EV therapeutic, especially for diseases with no existing treatment.
This report adds to a growing body of literature demonstrating the potential - and shortcomings - of targeted EV Therapeutics. We believe that future improvements in targeting specificity, DNA packaging and EV isolation would open new perspectives into EV-mediated therapeutic delivery as a safe and effective technology.
Conclusion
In conclusion, our engineered EV generation technique is simple, robust, and efficient. This study demonstrates that small peptide-based ligands can impart affinity to EVs upon being displayed on the surface. EV-mediated targeted delivery was achieved without any observed toxicity in the cell lines or visible side effects on the mice. We believe that the EV-mediated targeted delivery can be leveraged for treating human pancreatic diseases. Moreover, conjugating therapeutic molecules/drugs/imaging probes with engineered EVs can be applied for investigating targeted delivery in other clinically significant organs. To tap into the tremendous potential of EV-mediated targeted delivery, further in-vivo research is needed to improve the pharmacokinetic profile of delivery systems and minimizing non-specific uptake of EVs.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Jeremy Hix, Dr. Alicia Withrow and the MSU Center for Advanced Microscopy for technical assistance in in vivo imaging IVIS, TEM, and confocal imaging. This work was supported by the startup fund for Dr. Harada provided by Michigan State University, and the James and Kathleen Cornelius endowment (Contag).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI. et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066
2. Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Springer. 2016
3. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255-89
4. Bobrie A, Colombo M, Krumeich S, Raposo G, Thery C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J Extracell Vesicles. 2012 1
5. Mork M, Andreasen JJ, Rasmussen LH, Lip GYH, Pedersen S, Baek R. et al. Elevated blood plasma levels of tissue factor-bearing extracellular vesicles in patients with atrial fibrillation. Thromb Res. 2019;173:141-50
6. Wu CX, Liu ZF. Proteomic Profiling of Sweat Exosome Suggests its Involvement in Skin Immunity. J Invest Dermatol. 2018;138:89-97
7. Somiya M, Yoshioka Y, Ochiya T. Biocompatibility of highly purified bovine milk-derived extracellular vesicles. J Extracell Vesicles. 2018;7:1440132
8. Hyun KA, Gwak H, Lee J, Kwak B, Jung HI. Salivary Exosome and Cell-Free DNA for Cancer Detection. Micromachines (Basel). 2018 9
9. Stahl PD, Raposo G. Exosomes and extracellular vesicles: the path forward. Essays Biochem. 2018;62:119-24
10. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213-28
11. Miao C, Chang J, Zhang G, Fang Y. MicroRNAs in type 1 diabetes: new research progress and potential directions. Biochem Cell Biol. 2018;96:498-506
12. Xu H, Jia S, Xu H. Potential therapeutic applications of exosomes in different autoimmune diseases. Clin Immunol. 2019;205:116-24
13. Garcia-Contreras M, Brooks RW, Boccuzzi L, Robbins PD, Ricordi C. Exosomes as biomarkers and therapeutic tools for type 1 diabetes mellitus. Eur Rev Med Pharmacol Sci. 2017;21:2940-56
14. Shigemoto-Kuroda T, Oh JY, Kim D-K, Jeong HJ, Park SY, Lee HJ. et al. MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem cell reports. 2017;8:1214-25
15. Pitt JM, Andre F, Amigorena S, Soria J-C, Eggermont A, Kroemer G. et al. Dendritic cell-derived exosomes for cancer therapy. The Journal of clinical investigation. 2016;126:1224-32
16. Tang XJ, Sun XY, Huang KM, Zhang L, Yang ZS, Zou DD. et al. Therapeutic potential of CAR-T cell-derived exosomes: a cell-free modality for targeted cancer therapy. Oncotarget. 2015;6:44179-90
17. Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H. et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Molecular therapy: the journal of the American Society of Gene Therapy. 2008;16:782-90
18. Doeppner TR, Herz J, Gorgens A, Schlechter J, Ludwig AK, Radtke S. et al. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl Med. 2015;4:1131-43
19. Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev. 2016;106:148-56
20. Kanada M, Bachmann MH, Hardy JW, Frimannson DO, Bronsart L, Wang A. et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci U S A. 2015;112:E1433-42
21. Zhu X, Badawi M, Pomeroy S, Sutaria DS, Xie Z, Baek A. et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6:1324730
22. Rountree RB, Mandl SJ, Nachtwey JM, Dalpozzo K, Do L, Lombardo JR. et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer research. 2011;71:5235-44
23. Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820:940-8
24. Samanta S, Rajasingh S, Drosos N, Zhou Z, Dawn B, Rajasingh J. Exosomes: new molecular targets of diseases. Acta Pharmacol Sin. 2018;39:501-13
25. Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal Stem Cell-derived Extracellular Vesicles: Toward Cell-free Therapeutic Applications. Mol Ther. 2015;23:812-23
26. Bian S, Zhang L, Duan L, Wang X, Min Y, Yu H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med (Berl). 2014;92:387-97
27. He J, Wang Y, Sun S, Yu M, Wang C, Pei X. et al. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology (Carlton). 2012;17:493-500
28. Kanada M, Kim BD, Hardy JW, Ronald JA, Bachmann MH, Bernard MP. et al. Microvesicle-Mediated Delivery of Minicircle DNA Results in Effective Gene-Directed Enzyme Prodrug Cancer Therapy. Mol Cancer Ther. 2019;18:2331-42
29. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185-91
30. Cooper JM, Wiklander PO, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M. et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Movement Disorders. 2014;29:1476-85
31. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology. 2011;29:341-5
32. Ingato D, Lee JU, Sim SJ, Kwon YJ. Good things come in small packages: Overcoming challenges to harness extracellular vesicles for therapeutic delivery. J Control Release. 2016;241:174-85
33. Gudbergsson JM, Jonsson K, Simonsen JB, Johnsen KB. Systematic review of targeted extracellular vesicles for drug delivery - Considerations on methodological and biological heterogeneity. J Control Release. 2019;306:108-20
34. Armstrong JP, Holme MN, Stevens MM. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano. 2017;11:69-83
35. Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release. 2015;219:396-405
36. Wiklander OP, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mäger I. et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316
37. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature biotechnology. 2011;29:341-5
38. Sedlik C, Vigneron J, Torrieri-Dramard L, Pitoiset F, Denizeau J, Chesneau C. et al. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. Journal of extracellular vesicles. 2014;3:24646
39. Yang J, Zhang X, Chen X, Wang L, Yang G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol Ther Nucleic Acids. 2017;7:278-87
40. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
41. Liang G, Kan S, Zhu Y, Feng S, Feng W, Gao S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int J Nanomedicine. 2018;13:585-99
42. Delcayre A, Estelles A, Sperinde J, Roulon T, Paz P, Aguilar B. et al. Exosome display technology: applications to the development of new diagnostics and therapeutics. Blood Cells, Molecules, and Diseases. 2005;35:158-68
43. Kooijmans SA, Gitz-Francois JJ, Schiffelers RM, Vader P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: a plug-and-play approach. Nanoscale. 2018;10:2413-26
44. Hartman ZC, Wei J, Glass OK, Guo H, Lei G, Yang X-Y. et al. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011;29:9361-7
45. Wang JH, Forterre AV, Zhao J, Frimannsson DO, Delcayre A, Antes TJ. et al. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol Cancer Ther. 2018;17:1133-42
46. Wan S, Zhang L, Wang S, Liu Y, Wu C, Cui C. et al. Molecular Recognition-Based DNA Nanoassemblies on the Surfaces of Nanosized Exosomes. J Am Chem Soc. 2017;139:5289-92
47. Morishita M, Takahashi Y, Nishikawa M, Sano K, Kato K, Yamashita T. et al. Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherin fusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J Pharm Sci. 2015;104:705-13
48. Burtea C, Laurent S, Crombez D, Delcambre S, Sermeus C, Millard I. et al. Development of a peptide-functionalized imaging nanoprobe for the targeting of (FXYD2) γa as a highly specific biomarker of pancreatic beta cells. Contrast Media Mol I. 2015;10:398-412
49. Demine S, Balhuizen A, Debaille V, Joosten L, Fereau M, Chilla SNM. et al. Imaging of human insulin secreting cells with Gd-DOTA-P88, a paramagnetic contrast agent targeting the beta cell biomarker FXYD2γa. Molecules. 2018;23:2100
50. Huang KC, Chen Z, Jiang Y, Akare S, Kolber-Simonds D, Condon K. et al. Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol Cancer Ther. 2016;15:1208-16
51. Zhuang M, Du D, Pu L, Song H, Deng M, Long Q. et al. SPION-Decorated Exosome Delivered BAY55-9837 Targeting the Pancreas through Magnetism to Improve the Blood GLC Response. Small. 2019;15:e1903135
52. Li PW, Luo S, Xiao LY, Tian BL, Wang L, Zhang ZR. et al. A novel gemcitabine derivative-loaded liposome with great pancreas-targeting ability. Acta Pharmacol Sin. 2019;40:1448-56
53. Zhang Y, Werling U, Edelmann W. Seamless ligation cloning extract (SLiCE) cloning method. DNA Cloning and Assembly Methods: Springer. 2014 p. 235-44
54. Motohashi K. A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis. BMC Biotechnology. 2015;15:47
55. Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM. et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nature biotechnology. 2011;29:73
56. Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357-69
57. Baker A, Saltik M, Lehrmann H, Killisch I, Mautner V, Lamm G. et al. Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene therapy. 1997;4:773-82
58. Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T. et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. Journal of biotechnology. 2013;165:77-84
59. Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology. 2006;30:3.22. 1-3. 9
60. Bosch S, De Beaurepaire L, Allard M, Mosser M, Heichette C, Chrétien D. et al. Trehalose prevents aggregation of exosomes and cryodamage. Sci Rep-Uk. 2016;6:36162
61. Hamaguchi K, Gaskins HR, Leiter EH. NIT-1, a pancreatic beta-cell line established from a transgenic NOD/Lt mouse. Diabetes. 1991;40:842-9
62. Isolation of plasmid DNA from mammalian cells using QIAprep kit. QIAGEN News 1995. p. 11.
63. Khemthongcharoen N, Ruangpracha A, Sarapukdee P, Rattanavarin S, Jolivot R, Jarujareet U. et al. Novel p16 binding peptide development for p16-overexpressing cancer cell detection using phage display. Journal of Peptide Science. 2015;21:265-73
64. Zhu X, Badawi M, Pomeroy S, Sutaria DS, Xie Z, Baek A. et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. Journal of extracellular vesicles. 2017;6:1324730
65. Li J, Chen X, Yi J, Liu Y, Li D, Wang J. et al. Identification and characterization of 293T cell-derived exosomes by profiling the protein, mRNA and microRNA components. PloS one. 2016 11
66. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of extracellular vesicles. 2018;7:1535750
67. Zhao H, Doyle TC, Wong RJ, Cao Y, Stevenson DK, Piwnica-Worms D. et al. Characterization of coelenterazine analogs for measurements of Renilla luciferase activity in live cells and living animals. Molecular Imaging. 2004;3:15353500200403181
68. Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Advanced drug delivery reviews. 2016;106:148-56
69. Salunkhe S, Dheeraj, Basak M, Chitkara D, Mittal A. Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: Strategies and significance. J Control Release. 2020;326:599-614
70. Flamez D, Roland I, Berton A, Kutlu B, Dufrane D, Beckers MC. et al. A genomic-based approach identifies FXYD domain containing ion transport regulator 2 (FXYD2)γa as a pancreatic beta cell-specific biomarker. Diabetologia. 2010;53:1372-83
71. Longatti A, Schindler C, Collinson A, Jenkinson L, Matthews C, Fitzpatrick L. et al. High affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles. Nanoscale. 2018;10:14230-44
72. Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A. et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228-35
73. Kooijmans SAA, Gitz-Francois J, Schiffelers RM, Vader P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: a plug-and-play approach. Nanoscale. 2018;10:2413-26
74. Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J. et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015;4:e1008371
75. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
76. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
77. Hyenne V, Lefebvre O, Goetz JG. Going live with tumor exosomes and microvesicles. Cell Adh Migr. 2017;11:173-86
78. Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. Journal of extracellular vesicles. 2014;3:24641
79. Ramirez MI, Amorim MG, Gadelha C, Milic I, Welsh JA, Freitas VM. et al. Technical challenges of working with extracellular vesicles. Nanoscale. 2018;10:881-906
80. Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145-55
81. Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C. et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18:1606-14
82. Rashid MH, Borin TF, Ara R, Angara K, Cai J, Achyut BR. et al. Differential in vivo biodistribution of. Nanomedicine. 2019;21:102072
83. Faruqu FN, Wang JT, Xu L, McNickle L, Chong EM, Walters A. et al. Membrane Radiolabelling of Exosomes for Comparative Biodistribution Analysis in Immunocompetent and Immunodeficient Mice - A Novel and Universal Approach. Theranostics. 2019;9:1666-82
84. Amorim MG, Valieris R, Drummond RD, Pizzi MP, Freitas VM, Sinigaglia-Coimbra R. et al. A total transcriptome profiling method for plasma-derived extracellular vesicles: applications for liquid biopsies. Sci Rep. 2017;7:14395
85. Yang Z, Shi J, Xie J, Wang Y, Sun J, Liu T. et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat Biomed Eng. 2020;4:69-83
Author contact
![]() Corresponding author: Masako Harada, Ph.D., 4114 Institute for Quantitative Health Science and Engineering (IQ), Michigan State University, 775 Woodlot Dr, East Lansing, Michigan. Email: masharedu; Phone: 517-884-6940.
Corresponding author: Masako Harada, Ph.D., 4114 Institute for Quantitative Health Science and Engineering (IQ), Michigan State University, 775 Woodlot Dr, East Lansing, Michigan. Email: masharedu; Phone: 517-884-6940.