- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods to track cells in vivo...
Labeling of cells with...
Maintenance of immune cell...
Monitoring of transferred cells...
Monitoring of transferred cells...
Pitfalls and clinical...
Concluding remark
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2021; 5(1):27-35. doi:10.7150/ntno.51391 This issue Cite
Review
Imaging of cell-based therapy using 89Zr-oxine ex vivo cell labeling for positron emission tomography
Yutaka Kurebayashi, Peter L. Choyke, Noriko Sato ![]()
Molecular Imaging Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Received 2020-8-1; Accepted 2020-10-13; Published 2021-1-1
Abstract

With the rapid development of anti-cancer cell-based therapies, such as adoptive T cell therapies using tumor-infiltrating T cells, T cell receptor transduced T cells, and chimeric antigen receptor T cells, there has been a growing interest in imaging technologies to non-invasively track transferred cells in vivo. Cell tracking using ex vivo cell labeling with positron emitting radioisotopes for positron emission tomography (PET) imaging has potential advantages over single-photon emitting radioisotopes. These advantages include intrinsically higher resolution, higher sensitivity, and higher signal-to-background ratios. Here, we review the current status of recently developed Zirconium-89 (89Zr)-oxine ex vivo cell labeling with PET imaging focusing on its applications and future perspectives. Labeling of cells with 89Zr-oxine is completed in a series of relatively simple steps, and its low radioactivity doses required for imaging does not interfere with the proliferation or function of the labeled immune cells. Preclinical studies have revealed that 89Zr-oxine PET allows high-resolution in vivo tracking of labeled cells for 1-2 weeks after cell transfer both in mice and non-human primates. These results provide a strong rationale for the clinical translation of 89Zr-oxine PET-based imaging of cell-based therapy.
Keywords: Zirconium-89, Zirconium-89 oxine, cell tracking, positron emission tomography, cell-based therapy
Introduction
The last decades of development in cell-based anti-cancer therapies has been marked by breakthroughs such as adoptive transfer of tumor-infiltrating T cells (TIL) [1, 2], tumor antigen specific T cell receptor (TCR)-transduced T cells (TCR-T) [3, 4], and chimeric antigen receptor T (CAR-T) cells [5-9]. Adoptive transfer of TIL in melanoma patients achieves objective responses of 50 - 70 % and complete responses of 22 % when a lymphodepleting preparative regimen is employed before transfer [1, 2, 10, 11]. CD19-directed CAR-T cells achieve durable complete responses in 40 % of refractory diffuse large B cell lymphoma and in 70 % of follicular lymphoma. However, some severe, sometimes fatal, side effects, including cytokine release syndrome that correlates with tumor burden and encephalopathy, have been observed [9, 12]. Along with the evolution of CAR-T cells with improved intracellular signal transduction to enable proliferation and cytokine production of T cells [9], CAR technology has been expanded to other cell types such as B cells and NK cells [13]. Other major breakthroughs have been made in the field of stem cell therapies. In addition to traditional hematopoietic stem cell transplants, mesenchymal stem cells or cells differentiated from induced pluripotent stem cell (iPSC) developed from the patient's own cells or embryonic stem cells have been actively investigated for the therapy or regeneration of damaged tissue [14, 15].
Common to all the cell-based therapies is their dependency on the delivery of transferred cells to the desired target in order for the cells to exert therapeutic effects. Even in the case of CAR-T cells that have enhanced target antigen recognition capabilities, the cells first have to migrate to and then infiltrate the target (e.g. cancer) tissue in order for them to recognize the specific antigen and exert cytotoxicity [16-18]. The accumulation of transferred tumor-targeting lymphocytes can vary among the metastatic lesions within the same patient as a result of different evolutionary and/or immunoediting processes unique to each metastatic lesion [19, 20]. In most clinical cell-based therapies, biopsies or observations of clinical outcome have been the method to evaluate the migration of transferred cells to the target tissue. Even if tumors are biopsied, transferred lymphocytes often cannot be distinguished from endogenous tumor-infiltrating lymphocytes unless transferred cells are labeled in advance [21, 22]. The number of cells reaching the target are often quite small. Therefore, imaging of transferred cells is clinically important in order to non-invasively and quantitatively evaluate the homing of the cells to the targeted cancers but also to use as a tool in designing next generation cell therapies with better homing characteristics.
Technically, transferred cells can be tracked using single-photon emission computed tomography (SPECT) or positron emission tomography (PET) when the cells are labeled with appropriate radioisotopes. Indium-111 (111In)-oxine or Technetium-99m (99mTc)-hexamethylpropylene amine oxime (HMPAO) scintigraphy/SPECT has been a classical method to visualize leukocytes and is clinically used for evaluation of inflammation, infection or abscess since the mid-1970s [23-26]. 111In-oxine scintigraphy/SPECT has been previously used to analyze the distribution of adoptively transferred TILs in patients [21, 22]. However, the sensitivity of SPECT is 2 to 3 orders of magnitude lower than that of PET [27], and relatively high doses of radioactivity are required for imaging, which cause death or dysfunction in the labeled cells [28-30]. 99mTc has a short half-life (6 hours) and is not suitable for tracing the transferred cells for multiple days, and cell labeling with 99mTc-HMPAO is relatively unstable [30, 31]. Alternatives to radiolabeling include magnetic resonance imaging (MRI) using superparamagnetic iron oxide nanoparticles (SPION) that are phagocytized by the cells to be tracked [32]. While MRI provides detailed anatomical information without ionizing radiation, it is cumbersome to survey the entire body and also difficult to detect the signal loss caused by iron oxide unless pre- and post-SPION loading studies are compared. This poses practical concerns as well. Moreover, quantitation of SPION's dark signal void is difficult. One of the recent developments in the tracking of cells with MRI is to use Fluorine-19 (19F) labeling. Here, the MRI unit is tuned to the resonance frequency of 19F, which blocks out all the background signal from water protons. However, this method requires specialized 19F detection coils, and because of limitations in sensitivity, large amounts of 19F have to be introduced to the cells or tissues to generate detectable signals [32, 33].
In general, PET is superior in image quality, sensitivity, spatial resolution, and quantification to SPECT [27], which makes it an attractive modality for tracking cells. Among positron (β)-emitting radioisotopes used for PET, 89Zr has a relatively long half-life (3.3 days), ideal for tracking cells for several days, and relatively low positron energy that is required for high resolution in PET [34, 35]. Therefore, PET imaging using 89Zr has been gaining attention, and the usefulness of 89Zr-immunoPET, in which antibodies or antibody fragments are labeled with 89Zr (e.g. 89Zr-labeled anti-PD-1/PD-L1 antibody), for imaging human patients has been evaluated [36-39]. For the specific task of tracking adoptively transferred cells for multiple days with relatively high resolution, cells to be tracked are labeled with 89Zr-oxine and imaged by PET [40, 41]. Here, we briefly review the current status of cell labeling and the applications of 89Zr-oxine PET imaging technique for cell-based therapy.
Methods to track cells in vivo by PET imaging
Radionuclides that have been used for ex vivo cell labeling to longitudinally evaluate the distribution of the cells of interest in vivo include; Fluorine-18 (t1/2 = 109.7 minutes), in the form of 18F-fluorodeoxyglucose (FDG) to label cells ex vivo before transfer [42], Copper-64 (64Cu, t1/2 = 12.7 hours), such as copper-64-pyruvaldehyde-bis (N4-methylthiosemicarbazone) (64Cu-PTSM) [43], and 89Zr and Iodine-124 (t1/2 = 4.2 days), in the form of radio-labeled antibodies against specific cell surface proteins [44]. 18F-FDG depends on glucose uptake by the cell and thus is not suitable for dormant cells [45]. 18F-FDG also suffers from relatively rapid efflux after labeling, due to dephosphorylation of 18F-FDG-6-phosphate, the phosphorylated form of 18F-FDG that will be trapped within the cell [42, 45, 46]. 64Cu-SPION has been recently developed as a PET-MRI multi-modal imaging nanoparticle that has potential benefits for tracking cells in vivo, taking advantage of both the high sensitivity of PET and detailed anatomical information of MRI [47]. However, the half-life of 64Cu, although longer than that of 18F, is still insufficient for tracking cells beyond several days after transfer. On the other hand, 89Zr, with a half-life of 3.3 days, is ideal for cell tracking for 1-2 weeks. Its relatively lower positron energy improves PET spatial resolution and thus, is most often used for studies of this type.
In in vivo cell labeling, a radioactive labeling reagent (e.g. radioisotope-conjugated antibodies, antibody fragments, or engineered antibodies for immunoPET) is systemically administered and the distribution of the target molecule expressed on the endogenous cell populations in the body is evaluated [36-39]. Thus, in vivo labeling cannot discriminate adoptively transferred cells from the endogenous cells expressing the same target molecules (e.g. adoptively transferred CD8 T cell versus endogenous CD8 T cells). Therefore, although in vivo cell labeling is useful for analyzing the distribution of endogenous cell populations [38, 39], it is not suitable for the evaluation of the fraction of adoptively transferred cells that reach the target tissue. In vivo labeling is frequently associated with high background signal due to specific or non-specific antibody distribution (e.g. liver and spleen) and clearance (e.g. renal clearance of antibody fragments), long circulation time in the blood and pooling in tissue fluid, including those caused by enhanced permeability and retention (EPR) effect in some tumor tissues [48]. In vivo labeling also requires highly specific antibodies with little to no cross-binding to non-target cells; however, specific cell markers, although present on many T and B cells, are frequently unavailable for many other cell types such as dendritic cells or monocytes.
If adoptively transferred cells are transduced with a PET reporter gene, it is possible to administer a radioactive reporter probe to locate the transferred cells in the body [49]. Since the expression level of the reporter gene is maintained after cell proliferation, reporter gene imaging can achieve longer-term tracking of transferred cells. It also detects only live cells. On the other hand, the requirement for an injection of the radioactive probe and clearance of unbound probe at each interested time point limits short-term or frequent monitoring of cell migration. Thus, monitoring cellular responses to additional stimuli that cause relatively rapid changes of cell distribution could be difficult. The biggest disadvantage of PET reporter gene imaging is its requirement for gene introduction to the cells. It is imperative that such reporter genes are carefully chosen to avoid immunogenicity. Although PET reporter gene imaging is feasible for therapeutic cells that undergo genetic engineering during their production, such as CAR-T cells, for cells that do not require genetic engineering, which are the majority of TILs and stem cells, reporter gene imaging strategies would be difficult to translate into the clinic.
In contrast to in vivo labeling, cells can be labeled with radioisotopes ex vivo before adoptive transfer to patients [40, 41]. Since only the cells adoptively transferred are labeled with radioisotope, ex vivo labeling has essentially zero background signal in the recipient, thereby achieving extremely high signal-to-background ratios even with low cell labeling doses, and, therefore, high sensitivity to even tiny clusters of labeled cells [40, 41]. Monitoring relatively small cell distribution changes to additional stimuli could be possible with this method. Ex vivo labeling can accurately track adoptively transferred cells. However, some background signals may develop over time due to phagocytosis of damaged labeled cells or release of the radioactive label after death of the labeled cells (e.g. accumulation of 89Zr within bone as described below). As each cell division results in approximately halving of the label per cell, image intensity can decrease due to cell division alone [40].
Labeling of cells with 89Zr-oxine for PET imaging
Cell labeling by 89Zr-oxine is completed in three relatively simple steps that can be performed in aqueous solution [40]: 1) synthesis of 89Zr-oxine by mixing 89Zr-chloride with oxine followed by neutralization at room temperature. 89Zr-chloride can be generated from 89Zr-oxalate produced by a cyclotron [50], 2) incubation of cells with synthesized 89Zr-oxine for 15 minutes at room temperature or at 4 oC, and 3) washing of the labeled cells. The 89Zr-oxine synthesis yield is as high as >98 %, which, together with the aqueous conditions, eliminates the need for a purification step. With application of the generated 89Zr-oxine solution to a cell suspension, virtually all the cells are labeled with 89Zr-oxine. The synthesis and labeling steps are similar to those of 111In-oxine complex and thus, 89Zr-oxine cell labeling could be easily translated to the clinic. Recently, a slightly modified method of generation of 89Zr-oxine, which uses 89Zr-oxalate instead of 89Zr-chloride, has been reported [51]. Another reported method of synthesis uses neutralized 89Zr-oxalate to react with oxine in chloroform, followed by recovery of the synthesized 89Zr-oxine from chloroform phase by evaporation and re-dissolving the obtained 89Zr-oxine in dimethyl sulfoxide for further dilution in aqueous buffer for cell labeling [40, 41].
Mechanistically, the lipid solubility of 89Zr-oxine allows it to diffuse through the cell membrane. Therefore, labeling with 89Zr-oxine does not require active uptake by cells and can be performed in resting cells and at 4 oC [40]. This characteristic also eliminates the requirement for expression of certain cell surface molecules for labeling or an antibody specific cell surface protein.
Experience with 111In-oxine is instructive. 8-hydroxyquinoline (oxine) can be released from 111In-oxine after the molecule enters the cytoplasm, and the free 111In forms a stable complex with cytoplasmic and nuclear proteins [52]. The formation of 111In-protein complex is presumably achieved through exchange of 111In from 8-hydroxyquinoline to intracellular proteins [52]. Free 8-hydroxyquinoline is then exported from the cell. Although a similar mechanism can be assumed for 89Zr-oxine, the exact mechanism of cell labeling using 89Zr-oxine is still unclear. Our preliminary results have shown that 89Zr-oxine also labels the cell membranes (cytoplasmic and intracellular membranes) as well as cytoplasmic, and nuclear proteins, while negligible binding was observed with chromatin and cytoskeleton proteins (unpublished data). Although 89Zr-oxine can label a variety of cells, the labeling efficiency (percentage of 89Zr incorporated to the cells per added activity) differs among cell types [40]. This may be due to the difference in the amount of intra-cellular proteins available for 89Zr binding. 89Zr-oxine labeled cells, especially non-dividing cells (e.g. matured dendritic cells), retain 89Zr very well over multiple days [40].
In contrast to the cell-permeable 89Zr-oxine, 89Zr-deferoxamine-NCS has been reported to directly conjugate 89Zr to cell surface proteins for cell tracking by PET [53, 54]. In this method, deferoxamine chelates 89Zr, while the NCS group covalently binds primary amine groups of membrane proteins on the cell surface. Therefore, 89Zr radioactivity is mainly detected in the membrane fraction of labeled cells but not in cytoplasmic and nuclear fractions [53].
Maintenance of immune cell function after labeling with 89Zr-oxine
Importantly, labeling of immune cells including lymphocytes, macrophages, and bone marrow-derived dendritic cells with 89Zr-oxine does not affect their function when optimal labeling doses were used. For example, labeling with 89Zr-oxine does not interfere with the activation, proliferation, and cytokine expression (IFN-γ and IL-2) after TCR stimulation in labeled CD8 T cells [40]. Similarly, labeling of CAR-T cells by 89Zr-oxine does not affect their viability, cytotoxicity, or cytokine production [51]. Labeling with 89Zr-oxine does not interfere with LPS-induced activation of dendritic cells and their ability to present antigens and activate T cells [40]. The only exception observed to date, is that 89Zr-oxine labeling of bone marrow cells, even at low radioactivity doses (10.9 kBq/106 cells), showed initial delay in the proliferation after granulocyte colony stimulation factor (GM-CSF) stimulation in vitro [55]. However, the differentiation capacity of these labeled bone marrow cells is maintained both in vitro and in vivo [55]. Based on these studies, the optimal labeling doses minimize cellular toxicity while being sufficient to image by PET are approximately 11 - 44 kBq/106 cells, depending on type and condition of the cells.
Once labeled with 89Zr-oxine, 89Zr is stably retained in labeled cells [40]. However, death of labeled cells results in rapid release of free 89Zr. In vivo, free 89Zr is excreted from the kidney; however, a small fraction of free 89Zr may be taken up in bone matrix hydroxyapatite [40, 55, 56]. This small uptake in bone can be reduced by administration of deferoxamine, a chelating agent clinically used for treatment of iron overload and hemochromatosis [55]. Once bound to deferoxamine, the 89Zr is rapidly excreted in the urinary tract.
Monitoring of transferred cells using 89Zr-oxine PET in mouse models
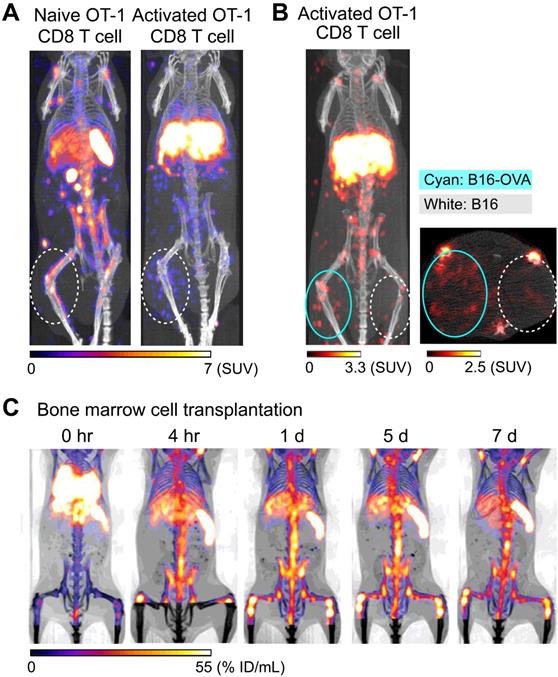
Preclinical studies have shown that 89Zr-oxine PET can track adoptively transferred cells over a period of 1-2 weeks. For example, adoptive transfer of 89Zr-oxine-labeled activated OT-I CD8 T cells (TCR transgenic T cells that recognize ovalbumin: OVA) shows initial rapid accumulation in the lungs, followed by gradual accumulation within antigen-expressing tumor (B16-OVA) by Day 2 (Figure 1A, right) [40]. In contrast, these activated OT-I CD8 T cells do not accumulate in antigen-negative B16 tumors. Uptake is restricted to B16-OVA tumors, which is associated with a rapid anti-tumor effect (Figure 1B). In addition to the antigen-expressing B16-OVA tumor, 89Zr-oxine PET shows that activated OT-I CD8 T cells also distribute to the liver after adoptive transfer, compatible with previous observations that activated CD8 T cells preferentially home to the liver [57, 58]. On the other hand, 89Zr-oxine PET demonstrates that naïve CD8 T cells preferentially distribute to the spleen and lymph nodes instead of the liver (Figure 1A, left). Therefore, 89Zr-oxine PET clearly captures the different behaviors between naïve and activated CD8 T cells after adoptive transfer. Similar results have been observed when 89Zr-oxine-labeled CAR-T cells are used, wherein transferred prostate stem cell antigen (PSCA)-targeting CAR-T cells accumulate in the liver as well as the PSCA-expressing tumor, after initial distribution to the lungs [51].
Tracking of transferred cells using 89Zr-oxine PET in murine models. A) Activated OT-1 CD8 T cells distribute to antigen-expressing B16-OVA tumor (white dashed circle), while naïve OT-1 CD8 T cells do not. Naïve OT-1 CD8 T cells (left) and activated OT-1 CD8 T cells (right, adapted from [40]) were labeled with 89Zr-oxine (222 kBq/8 x 106 cells and 248.5 kBq/7.7 x 106 cells, respectively) and intravenously transferred to tumor-bearing recipient mice. PET/CT images acquired on 2nd day after transfer are shown. Also note the different distribution between naïve and activated OT-1 CD8 T cells outside the tumor. B) Activated OT-1 CD8 T cells distribute to antigen-expressing B16-OVA tumor (cyan circle) but not to antigen-negative B16 tumor (white dashed circle). Activated OT-1 CD8 T cells labeled with 89Zr-oxine (185 kBq/8 x 106 cells) were intravenously transferred to the recipient. MicroPET/CT images acquired on 4th day after transfer are shown. Left: maximum intensity projection MIP PET/CT, right: transverse plane. C) Distribution of transferred bone marrow cells. Donor bone marrow cells were labeled with 89Zr-oxine (16.6 kBq/2.0 x 107 cells) and intravenously transferred to the recipient. PET/CT imaging was performed at indicated time points after transfer, with multiple intramuscular injections of deferoxamine to prevent accumulation of free 89Zr within bone. Transferred bone marrow cells initially distribute to the lungs, followed by distribution within 4 hours to the bone marrow, liver, and spleen. Adapted from [55].
Vγ2Vδ9 subtype of γδ T cells, which represent most circulating γδ T cells in humans, can recognize phosphoantigens expressed on tumor cells and exert anti-tumor activity [59]. When ex vivo expanded human Vγ2Vδ9 T cells are labeled with 89Zr-oxine and transferred to tumor-bearing SCID mice, 89Zr-oxine PET shows accumulation in the tumor and liver, after transient distribution to the lungs. [60]. This can be detected by 89Zr-oxine PET and is augmented by administration of aminobisphosphonate, which increases the expression of phosphoantigens on the surface of tumors [59, 61].
Tracking of bone marrow cells also reveals interesting patterns. 89Zr-oxine PET shows the initial transient distribution of transferred bone marrow cells to the lungs, rapidly followed by homing to the bone marrow, spleen, and liver within 4 hours of transfer (Figure 1C) [55]. Since the homing of bone marrow cells is dependent on CXCL12-CXCR4 axis [62], 89Zr-oxine PET shows delayed homing of labeled donor bone marrow cells to recipient bone marrow after administration of plerixafor, a CXCR4 antagonist [55]. Co-administration of plerixafor and G-CSF further disturbed this homing process [55]. On the other hand, 89Zr-oxine PET shows that the initial homing of transferred bone marrow cells is not affected by prior whole body irradiation [55]. In a bone fracture model, 89Zr-oxine PET further shows the mobilization of pre-transferred 89Zr-labeled bone marrow cells from pre-distributed organs (bone marrow, spleen, and liver) to a site of fracture within 1 day of injury [63]. This re-distribution of pre-distributed bone marrow cells is also impaired by plerixafor treatment [63]. These reports indicate that 89Zr-oxine labeling of highly radiosensitive bone marrow could be performed with minimum effect on functionality of the cells.
With the 89Zr-deferoxamine-NCS direct membrane protein conjugation method, human mesenchymal stem cells have been imaged, showing their distribution to the lungs and liver after intravenous injection in athymic nude mice [53]. Interestingly most human mesenchymal stem cells labeled with 89Zr-deferoxamine-NCS remain within the injected site for at least one week after injection into myocardium in athymic nude mice [53].
Monitoring of transferred cells using 89Zr-oxine PET in non-human primates
89Zr-oxine PET can be used to image adoptively transferred cells in non-human primates. For example, in an autologous intravenous hematopoietic stem cell transplantation model in rhesus macaques, 89Zr-oxine PET imaging shows that CD34+ hematopoietic stem and progenitor cells (HSPCs) accumulate in the bone marrow, liver, and spleen after rapid passage through the lungs (Figure 2A) [64], similar to the pattern seen in a murine model of bone marrow transplantation (Figure 1C) [55]. Clinically, intrabone transplantation of HSPCs, especially cord blood cells, has been suggested as a method to improve engraftment in the recipient's bone marrow [65, 66], and preclinical studies using swine and macaque were undertaken to determine if homing and engraftment of HSPCs was improved with intrabone transplantation compared to intravenous transfer [67, 68]. 89Zr-oxine PET was used to further confirm the retention of HSPCs at the injection site in the iliac bone in rhesus macaques [69].
Tracking of transferred cells using 89Zr-oxine PET in non-human primate models. A) Distribution of transferred CD34+ hematopoietic stem and progenitor cells (HSPCs) in a rhesus macaque. CD34+ HSPCs mobilized by plerixafor and G-CSF treatment were labeled with 89Zr-oxine and autologously transferred intravenously (44.4 kBq/106 cells, 1.2 x 106 cells/kg). PET/CT was performed longitudinally under continuous infusion of deferoxamine to prevent accumulation of free 89Zr within bone. Transferred cells rapidly distribute to the bone marrow, liver, and spleen [64]. B) Distribution of transferred NK cells in a rhesus macaque. NK cells purified from peripheral blood were expanded ex vivo with IL-2, labeled with 89Zr-oxine, and autologously transferred intravenously (15.2 kBq/106 cells, 21.9 x 106 cells/kg). Cell migration was longitudinally tracked by PET/CT. Deferoxamine was continuously infused during the entire imaging study. Transferred NK cells initially distribute to the lungs, followed by gradual distribution to the liver and spleen. Little distribution of NK cells to the bone marrow is observed. Adapted from [64].
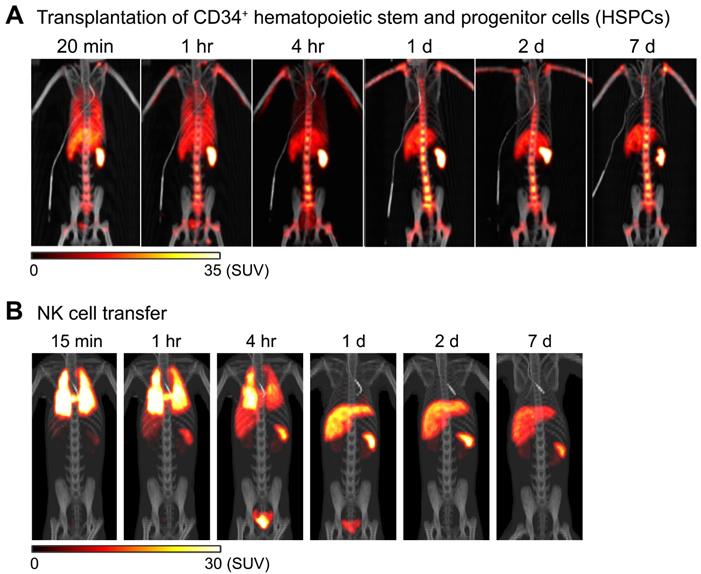
In another study, NK cells were expanded ex vivo with IL-2, per protocol used for human NK cell clinical trials, labeled with 89Zr-oxine, and tracked by 89Zr-oxine PET after autologous adoptive transfer, for preclinical evaluation of NK cell therapy against hematological malignancies such as leukemia. In this study, 89Zr-oxine PET revealed that transferred NK cells showed only minimal homing to the bone marrow, and most transferred NK cells distribute to the liver and spleen (Figure 2B) [64]. This implies that for NK cells to be useful in treating bone marrow malignancies, NK cells must be engineered to track to the bone marrow in greater numbers than unmodified NK cells.
In humans, it has been known since the 1940s that glucocorticoid treatment rapidly decreases the number of eosinophils in peripheral blood within 1-2 hours of administration [70]; however, it has been unclear where the eosinophils re-distribute in the body. To address this question, eosinophils purified from peripheral blood were labeled with 89Zr-oxine, transferred back to the animal, and imaging was performed before and after glucocorticoid treatment. 89Zr-oxine PET demonstrated a rapid increase of bone marrow accumulation of 89Zr-oxine labeled eosinophils over a four-hour period after glucocorticoid treatment [71]. This demonstrates the high sensitivity of 89Zr-oxine PET to detect rapid changes in cell distribution in response to exogenous stimuli.
These results indicate that high-resolution cell tracking using 89Zr-oxine PET can also be successfully performed in non-human primates using clinical PET scanners, providing a rationale for the translation of cell tracking using 89Zr-oxine PET to human trials.
Pitfalls and clinical translation of 89Zr-oxine PET
There are several pitfalls associated with 89Zr-oxine PET and other ex vivo labeling methods. One is the possible transfer of the label to phagocytes after the death of labeled cells. As observed in the use of iron nanoparticles for MRI [72], radioisotopes that are tightly bound to a cell structure or deposited in tissue could be transferred to tissue monocytes/macrophages by phagocytosis, resulting in non-specific signals. In this regard, studies using 89Zr-oxine so far have shown that 89Zr is released from dead cells and mostly cleared from the kidney. This process can be expedited by utilizing deferoxamine infusion as described below [55, 64]. 89Zr-oxine does not passively transfer from labeled cells to neighboring cells [64].
Another pitfall is that there can be bone uptake from small amounts of released 89Zr after death of labeled cells [40, 55, 56]. However, this can be again avoided by deferoxamine to chelate free 89Zr and enhance clearance from kidneys [55, 64]. Deferoxamine is clinically used for iron overdose and thus potentially could be used safely during the imaging.
Before translation of 89Zr-oxine PET technology into the clinical setting, a number of important criteria must be met. There must be simplicity in the labeling method, low cytotoxicity in labeled cells, and no side effects to recipients of labeled cells. Cell labeling with 89Zr-oxine is straightforward and exerts minimal toxicity to labeled cells when optimal cell labeling doses were used. Although no clinical trials in humans have been performed, dosimetry data in non-human primates that received autologous NK cells showed very low radio-exposures to organs [64], and no clinical side effects were observed with autologous transfer of NK cells, CD34+ HPSCs [64] and eosinophils [71], confirming the safety of this method. Practically, labeling only a small fraction (e.g. 10 %) of the cells infused for cell-based therapy should be sufficient to obtain PET images with sufficiently high quality for cell tracking.
Concluding remark
Here, we have reviewed our experience with cell labeling using 89Zr-oxine in preclinical models of in vivo tracking. Cell labeling with 89Zr-oxine can be performed in relatively simple steps. Once labeled, 89Zr is stably retained in cells until cell death. After cell death, 89Zr is quickly liberated and generally cleared from the kidneys with enhanced clearance if deferoxamine is infused. 89Zr-oxine PET allows high-resolution tracking of transferred cells both in murine and non-human primate models for 1-2 weeks after transfer. These preclinical observations collectively provide a strong rationale for clinical translation of this technology.
Abbreviations
CAR-T: chimeric antigen receptor T; HSPC: hematopoietic stem and progenitor cell; iPSC: induced pluripotent stem cell; MRI: magnetic resonance imaging; NK: natural killer; OVA: ovalbumin; PET: positron emission tomography; SPECT: single photon emission computed tomography; SPIO: superparamagnetic iron oxide; TCR: T cell receptor; TIL: tumor-infiltrating lymphocyte; 89Zr: Zirconium-89.
Competing interests
N.S. and P.L.C. are co-inventors on a U.S. patent for cell labeling using 89Zr-oxine technology and filed U.S. divisional and International patent applications for the synthesis and application of 89Zr-oxine complex.
References
1. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ. et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850-4
2. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299-308
3. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126-9
4. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917-24
5. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720-4
6. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188:619-26
7. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C. et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279-86
8. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38
9. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361-5
10. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550-7
11. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U. et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233-9
12. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O. et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377:2545-54
13. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R. et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382:545-53
14. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16:115-30
15. Kimbrel EA, Lanza R. Next-generation stem cells — ushering in a new era of cell-based therapies. Nature Reviews Drug Discovery. 2020;19:463-79
16. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol. 2019;10:128
17. Kosti P, Maher J, Arnold JN. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front Immunol. 2018;9:1104
18. Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol. 2018;9:2593
19. Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA. et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell. 2017;170:927-38 e20
20. Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L. et al. Evolution of Metastases in Space and Time under Immune Selection. Cell. 2018;175:751-65 e16
21. Griffith KD, Read EJ, Carrasquillo JA, Carter CS, Yang JC, Fisher B. et al. In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J Natl Cancer Inst. 1989;81:1709-17
22. Fisher B, Packard BS, Read EJ, Carrasquillo JA, Carter CS, Topalian SL. et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. 1989;7:250-61
23. McAfee JG, Thakur MLJJonmop, Society of Nuclear Medicine. Survey of radioactive agents for in vitro labeling of phagocytic leukocytes. I. Soluble agents. J Nucl Med. 1976;17:480-7
24. Segal A, Arnot R, Thakur M, Lavender JJTL. Indium-111-labelled leucocytes for localisation of abscesses. Lancet. 1976;308:1056-8
25. Segal A, Munro JM, Ensell J, Sarner MJTL. Indium-111 tagged leucocytes in the diagnosis of inflammatory bowel disease. Lancet. 1981;318:230-2
26. de Vries EF, Roca M, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with (99m)Tc-HMPAO. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. 2010;37:842-8
27. Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193-207
28. Bulte JWM, Daldrup-Link HE. Clinical Tracking of Cell Transfer and Cell Transplantation: Trials and Tribulations. Radiology. 2018;289:604-15
29. Nowak B, Weber C, Schober A, Zeiffer U, Liehn EA, von Hundelshausen P. et al. Indium-111 oxine labelling affects the cellular integrity of haematopoietic progenitor cells. Eur J Nucl Med Mol Imaging. 2007;34:715-21
30. Botti C, Negri DR, Seregni E, Ramakrishna V, Arienti F, Maffioli L. et al. Comparison of three different methods for radiolabelling human activated T lymphocytes. Eur J Nucl Med. 1997;24:497-504
31. Blocklet D, Toungouz M, Kiss R, Lambermont M, Velu T, Duriau D. et al. 111In-oxine and 99mTc-HMPAO labelling of antigen-loaded dendritic cells: in vivo imaging and influence on motility and actin content. Eur J Nucl Med Mol Imaging. 2003;30:440-7
32. Ahrens ET, Bulte JW. Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol. 2013;13:755-63
33. Chapelin F, Capitini CM, Ahrens ET. Fluorine-19 MRI for detection and quantification of immune cell therapy for cancer. J Immunother Cancer. 2018;6:105
34. Deri MA, Zeglis BM, Francesconi LC, Lewis JS. PET imaging with (8)(9)Zr: from radiochemistry to the clinic. Nucl Med Biol. 2013;40:3-14
35. Conti M, Eriksson L. Physics of pure and non-pure positron emitters for PET: a review and a discussion. EJNMMI Phys. 2016;3:8
36. Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC. et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. 2018;24:1852-8
37. Wei W, Rosenkrans ZT, Liu J, Huang G, Luo QY, Cai W. ImmunoPET: Concept, Design, and Applications. Chem Rev. 2020;120:3787-851
38. Pandit-Taskar N, Postow MA, Hellmann MD, Harding JJ, Barker CA, O'Donoghue JA. et al. First-in-Humans Imaging with (89)Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J Nucl Med. 2020;61:512-9
39. Tavare R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB. et al. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res. 2016;76:73-82
40. Sato N, Wu H, Asiedu KO, Szajek LP, Griffiths GL, Choyke PL. (89)Zr-Oxine Complex PET Cell Imaging in Monitoring Cell-based Therapies. Radiology. 2015;275:490-500
41. Charoenphun P, Meszaros LK, Chuamsaamarkkee K, Sharif-Paghaleh E, Ballinger JR, Ferris TJ. et al. [(89)Zr]oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur J Nucl Med Mol Imaging. 2015;42:278-87
42. Grabner A, Kentrup D, Edemir B, Sirin Y, Pavenstadt H, Schlatter E. et al. PET with 18F-FDG-labeled T lymphocytes for diagnosis of acute rat renal allograft rejection. J Nucl Med. 2013;54:1147-53
43. Adonai N, Adonai N, Nguyen KN, Walsh J, Iyer M, Toyokuni T. et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci U S A. 2002;99:3030-5
44. Zettlitz KA, Tavare R, Knowles SM, Steward KK, Timmerman JM, Wu AM. ImmunoPET of Malignant and Normal B Cells with (89)Zr- and (124)I-Labeled Obinutuzumab Antibody Fragments Reveals Differential CD20 Internalization In Vivo. Clin Cancer Res. 2017;23:7242-52
45. Stojanov K, de Vries EF, Hoekstra D, van Waarde A, Dierckx RA, Zuhorn IS. [18F]FDG labeling of neural stem cells for in vivo cell tracking with positron emission tomography: inhibition of tracer release by phloretin. Mol Imaging. 2012;11:1-12
46. Ritchie D, Mileshkin L, Wall D, Bartholeyns J, Thompson M, Coverdale J. et al. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol Immunother. 2007;56:155-63
47. Bhatnagar P, Alauddin M, Bankson JA, Kirui D, Seifi P, Huls H. et al. Tumor lysing genetically engineered T cells loaded with multi-modal imaging agents. Sci Rep. 2014;4:4502
48. Matsumura Y, Maeda HJCr. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
49. Ashmore-Harris C, Iafrate M, Saleem A, Fruhwirth GO. Non-invasive Reporter Gene Imaging of Cell Therapies, including T Cells and Stem Cells. Mol Ther. 2020;28:1392-416
50. Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36:729-39
51. Weist MR, Starr R, Aguilar B, Chea J, Miles JK, Poku E. et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with (89)Zr-Oxine. J Nucl Med. 2018;59:1531-7
52. Roca M, de Vries EF, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with (111)In-oxine. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. 2010;37:835-41
53. Bansal A, Pandey MK, Demirhan YE, Nesbitt JJ, Crespo-Diaz RJ, Terzic A. et al. Novel (89)Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015;5:19
54. Lee SH, Soh H, Chung JH, Cho EH, Lee SJ, Ju JM. et al. Feasibility of real-time in vivo 89Zr-DFO-labeled CAR T-cell trafficking using PET imaging. PLoS One. 2020;15:e0223814
55. Asiedu KO, Koyasu S, Szajek LP, Choyke PL, Sato N. Bone Marrow Cell Trafficking Analyzed by (89)Zr-oxine Positron Emission Tomography in a Murine Transplantation Model. Clin Cancer Res. 2017;23:2759-68
56. Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89Zr in mice. Nucl Med Biol. 2011;38:675-81
57. John B, Crispe IN. Passive and active mechanisms trap activated CD8+ T cells in the liver. J Immunol. 2004;172:5222-9
58. Huang L, Soldevila G, Leeker M, Flavell R, Crispe IN. The liver eliminates T cells undergoing antigen-triggered apoptosis in vivo. Immunity. 1994;1:741-9
59. Silva-Santos B, Mensurado S, Coffelt SB. gammadelta T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat Rev Cancer. 2019;19:392-404
60. Man F, Lim L, Volpe A, Gabizon A, Shmeeda H, Draper B. et al. In Vivo PET Tracking of (89)Zr-Labeled Vgamma9Vdelta2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol Ther. 2019;27:219-29
61. Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 2000;96:384-92
62. Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901-10
63. Asiedu KO, Ferdousi M, Ton PT, Adler SS, Choyke PL, Sato N. Bone marrow cell homing to sites of acute tibial fracture: (89)Zr-oxine cell labeling with positron emission tomographic imaging in a mouse model. EJNMMI Res. 2018;8:109
64. Sato N, Stringaris K, Davidson-Moncada JK, Reger R, Adler SS, Dunbar C. et al. In Vivo Tracking of Adoptively Transferred Natural Killer Cells in Rhesus Macaques Using (89)Zirconium-Oxine Cell Labeling and PET Imaging. Clin Cancer Res. 2020;26:2573-81
65. Hagglund H, Ringden O, Agren B, Wennberg L, Remberger M, Rundquist L. et al. Intraosseous compared to intravenous infusion of allogeneic bone marrow. Bone Marrow Transplant. 1998;21:331-5
66. Frassoni F, Gualandi F, Podesta M, Raiola AM, Ibatici A, Piaggio G. et al. Direct intrabone transplant of unrelated cord-blood cells in acute leukaemia: a phase I/II study. Lancet Oncol. 2008;9:831-9
67. Feng Q, Chow PK, Frassoni F, Phua CM, Tan PK, Prasath A. et al. Nonhuman primate allogeneic hematopoietic stem cell transplantation by intraosseus vs intravenous injection: Engraftment, donor cell distribution, and mechanistic basis. Exp Hematol. 2008;36:1556-66
68. Pantin JM, Hoyt RF Jr, Aras O, Sato N, Chen MY, Hunt T. et al. Optimization of intrabone delivery of hematopoietic progenitor cells in a swine model using cell radiolabeling with [89]zirconium. Am J Transplant. 2015;15:606-17
69. Stringaris K, Hoyt RF Jr, Davidson-Moncada JK, Pantin JM, Tisdale JF, Uchida N. et al. Intrabone Transplantation of CD34+ cells with Optimized Delivery Does Not Enhance Engraftment in a Rhesus Macaque Model. Blood Advances. 2020 in press
70. Khoury P, Stokes K, Gadkari M, Makiya MA, Legrand F, Hu Z. et al. Glucocorticoid-induced eosinopenia in humans can be linked to early transcriptional events. Allergy. 2018;73:2076-9
71. Hong SG, Sato N, Legrand F, Gadkari M, Makiya M, Stokes K. et al. Glucocorticoid-induced eosinopenia results from CXCR4-dependent bone marrow migration. Blood. 2020; [Epub ahead of print. 2020 Jul 13]
72. Cianciaruso C, Pagani A, Martelli C, Bacigaluppi M, Squadrito ML, Lo Dico A. et al. Cellular magnetic resonance with iron oxide nanoparticles: long-term persistence of SPIO signal in the CNS after transplanted cell death. Nanomedicine (Lond). 2014;9:1457-74
Author contact
![]() Corresponding author: Noriko Sato, MD, PhD. Email: satonnih.gov. Phone: 240-858-3079, Fax: 301-402-3191. Email: satonnih.gov
Corresponding author: Noriko Sato, MD, PhD. Email: satonnih.gov. Phone: 240-858-3079, Fax: 301-402-3191. Email: satonnih.gov