- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusions
Materials and Methods...
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2018; 2(4):387-402. doi:10.7150/ntno.26969 This issue Cite
Research Paper
A Scalable Method for Squalenoylation and Assembly of Multifunctional 64Cu-Labeled Squalenoylated Gemcitabine Nanoparticles
Samantha T. Tucci1, Jai W. Seo2, Hamilton Kakwere2, Azadeh Kheirolomoom2, Elizabeth S. Ingham1, Lisa M. Mahakian1, Sarah Tam1, Spencer Tumbale2, Mo Baikoghli3, R. Holland Cheng3, Katherine W. Ferrara2 ![]()
1. Department of Biomedical Engineering, University of California Davis, Davis, California, 95616, USA.
2. Department of Radiology, Stanford University, Palo Alto, CA 94304, USA
3. Department of Molecular and Cellular Biology, University of California Davis, Davis, California, 95616, USA.
Received 2018-4-30; Accepted 2018-9-2; Published 2018-9-5
Abstract

Squalenoylation of gemcitabine, a front-line therapy for pancreatic cancer, allows for improved cellular-level and system-wide drug delivery. The established methods to conjugate squalene to gemcitabine and to form nanoparticles (NPs) with the squalenoylated gemcitabine (SqGem) conjugate are cumbersome, time-consuming and can be difficult to reliably replicate. Further, the creation of multi-functional SqGem-based NP theranostics would facilitate characterization of in vivo pharmacokinetics and efficacy.
Methods: Squalenoylation conjugation chemistry was enhanced to improve reliability and scalability using tert-butyldimethylsilyl (TBDMS) protecting groups. We then optimized a scalable microfluidic mixing platform to produce SqGem-based NPs and evaluated the stability and morphology of select NP formulations using dynamic light scattering (DLS) and transmission electron microscopy (TEM). Cytotoxicity was evaluated in both PANC-1 and KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-Cre) pancreatic cancer cell lines. A 64Cu chelator (2-S-(4-aminobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid, NOTA) was squalenoylated and used with positron emission tomography (PET) imaging to monitor the in vivo fate of SqGem-based NPs.
Results: Squalenoylation yields of gemcitabine increased from 15% to 63%. Cholesterol-PEG-2k inclusion was required to form SqGem-based NPs using our technique, and additional cholesterol inclusion increased particle stability at room temperature; after 1 week the PDI of SqGem NPs with cholesterol was ~ 0.2 while the PDI of SqGem NPs lacking cholesterol was ~ 0.5. Similar or superior cytotoxicity was achieved for SqGem-based NPs compared to gemcitabine or Abraxane® when evaluated at a concentration of 10 µM. Squalenoylation of NOTA enabled in vivo monitoring of SqGem-based NP pharmacokinetics and biodistribution.
Conclusion: We present a scalable technique for fabricating efficacious squalenoylated-gemcitabine nanoparticles and confirm their pharmacokinetic profile using a novel multifunctional 64Cu-SqNOTA-SqGem NP.
Keywords: Nanoparticles, gemcitabine, squalene, radiolabeling, 64Cu, pancreatic cancer
Introduction
Squalenoylation is a promising, novel technique that chemically modifies drug molecules, altering their systemic and cellular transport properties [1-6]. Squalenoylation requires a multistep organic synthesis to prepare squalene, a ubiquitous hydrocarbon and cholesterol precursor, for conjugation to a chemotherapeutic, siRNA, antibiotic, or other therapeutic moiety [1, 2, 7-13]. Squalenoylation offers particular promise for the delivery of gemcitabine, a front-line therapy for pancreatic ductal adenocarcinoma [1, 14-17]. Gemcitabine suffers from a short plasma half-life that can be enhanced by encapsulation. In addition, pancreatic tumors can quickly develop resistance to gemcitabine, and chemical modification of gemcitabine with squalene alters gemcitabine's transport on the cellular level, bypassing one mechanism of gemcitabine resistance [3, 18, 19]. Further, the squalenoylation of gemcitabine, a hydrophilic molecule, creates amphipathic chemicals capable of self-assembly into squalenoylated gemcitabine (SqGem)-based nanoparticles (NPs), which are cytotoxic in a variety of human tumor cell lines [15, 20]. The ability of SqGem to form NPs extends the plasma half-life and allows for active drug targeting, controlled drug release, and co-delivery of multiple therapeutic or imaging agents [3, 4, 8, 9, 17]. Here, in addition to improving upon existing methods of squalenoylation and formation of squalenoylated NPs, we evaluate the in vitro toxicity of SqGem-based NPs and evaluate their in vivo pharmacokinetics and organ distribution using positron emission tomographic (PET) imaging.
Although squalenoylation promises improvements in drug delivery, the fabrication of squalenoylated particles can be cumbersome and time-consuming, a result of both the squalenoylation procedure and the subsequent assembly of nanoparticles [1, 9]. Conjugation of squalene to form amphipathic molecules can be particularly challenging because the component molecules have opposing solubilities in typical solvent systems. These issues limit the reproducibility and eventual clinical translation of these promising therapeutics. In this report, we show that when tert-butyldimethylsilyl (TBDMS) protecting groups are used to increase gemcitabine's solubility in organic solvent systems, in a manner similar to gemcitabine conjugation to polymers [21-23], higher squalenoylation yields can be achieved.
SqGem-based NPs can be created using nanoprecipitation, a technique requiring precise control of numerous parameters, including the aqueous-to-organic solvent ratio, stir rate, droplet size, final solution surface area, and final solution volume. Even with precise control, manual nanoprecipitation techniques do not allow for scalable production of the resulting nanoassemblies. In our research, a commercially-available microfluidic device, the NanoAssemblr™ Benchtop, was optimized to produce reliably monodisperse SqGem-based NPs in a scalable manner and evaluated in conjunction with post-processing techniques for further SqGem-based NP refinement. Creating SqGem-based NPs using microfluidics is less cumbersome and error-prone than other bench-top methods. Microfluidic methods also facilitate investigations of alternate formulations by reducing nanoassembly variability.
SqGem-based NP toxicity was evaluated in vitro in both human (PANC-1) and murine (KPC, KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-Cre) pancreatic cancer cell lines and compared to free gemcitabine and Abraxane®, an FDA-approved nanotherapy for pancreatic cancer. The KPC cell line is derived from the genetically engineered KPC mouse model of pancreatic cancer, a model known to recapitulate the hypovascularity and dense stroma of human pancreatic cancer [24]. KPC cells can be cultured either as a monolayer or as organoids. When orthotopically implanting KPC organoids, the resulting tumors maintain their original phenotype; this phenotype is lost when implanting monolayer-cultured KPC cells [24]. SqGem-based NP efficacy differs with cell type and culturing technique but at high concentration is similar to or greater than free gemcitabine across multiple relevant pancreatic cancer models.
SqGem-based NPs have successfully cured or mitigated disease progress in multiple mouse models of cancer [6, 9, 16, 25], outperforming unmodified gemcitabine likely in part due to an enhanced pharmacokinetic profile [4]. Previously, the pharmacokinetic profile of SqGem-based NPs has been evaluated ex vivo using liquid chromatography-tandem mass spectrometry [4]. In vivo tracking of SqGem-based NPs has been limited to FRET (fluorescence resonance energy transfer) imaging of small animals and gadolinium-based contrast enhancement with MRI [10, 26-28]. PET imaging provides important insights in the field of controlled drug delivery, allowing quantitative in vivo assessment of biodistribution, local drug accumulation, and pharmacokinetics. We have previously developed methods to label liposomes and micelles with 64Cu using the 6-BAT chelator have shown these labels to be stable in serum over 48 hours [29]. Radiolabeling lipid-based NPs with 64Cu (half-life of 12.7 h) has been applied for quantitative tracking of the in vivo distribution of NPs over ~48 hours in multiple preclinical cancer models [30-37]. By squalenoylating a 64Cu chelator, 2-S-(4-aminobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), in vivo monitoring of squalene-based NPs is shown to be feasible and confirms a previous report for the early timepoints of the pharmacokinetic profile of SqGem-based NPs [7].
Below, we present the details of a scalable method to produce SqGem-based NPs, demonstrate their cytotoxicity in informative in vitro models of pancreatic cancer, and monitor SqGem-based NP behavior in vivo using PET imaging.
Results
Alternate synthesis methods increase yield of gemcitabine squalenoylation
Squalenoylation begins with the conversion of squalene (1) into 1,1',2-tris-norsqualenoyl acid (5, SqAcid) through a series of four chemical reaction steps (Scheme 1A). The Van Tamelen method, which favors action at terminal alkenes, first converts squalene into a mixture of halohydrins via oxidation with N-bromosuccinimide [2, 38, 39]. 2-Hydroxy-3-bromosqualene (2), the desired intermediate product, is isolated using column chromatography, providing a yield of 26%. Second, the single halohydrin on 2-hydroxy-3-bromosqualene (2) is converted into an epoxide group in the presence of a base, potassium carbonate. After extraction and washing, 2,3-oxidosqualene (3) remains at a high yield of approximately 97%. 2,3-Oxidosqualene (3) is then converted into 1,1',2-tris-norsqualene aldehyde (4), through the direct cleavage of the epoxide by periodic acid. After extraction and washing, 1,1',2-tris-norsqualene aldehyde (4) remains at a yield of 87%. In our experiments, we altered the methods of these first three steps slightly relative to those reported in the literature [9] by adjusting the reaction times, reaction scale (from 4.3 g to 17.16 g initial squalene), adding additional washes or extractions, and modifying column chromatography eluents. Conversion of squalene to 1,1',2-tris-norsqualene aldehyde has been successfully repeated in house ~20 times with similar yields.
Synthesis of SqGem (8). Conversion of squalene (1) to 1,1',2-Tris-norsqualenoyl acid (5) requires four steps with an overall yield of 11.4% (A). Protecting gemcitabine (6) with TBDMS allows for increased yields of SqGem (8) at the squalenoylation step, producing 63% compared to 15% in house replication (B) [9].
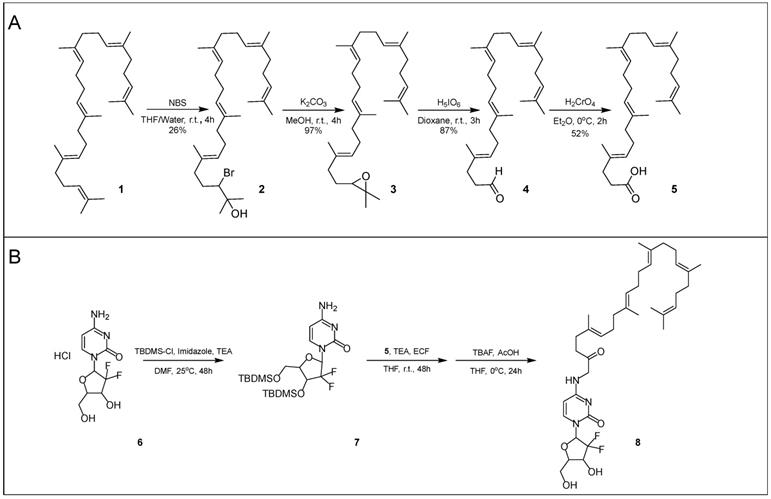
Next, 1,1',2-tris-norsqualene aldehyde (4) is oxidized into 1,1',2-tris-norsqualenoyl acid (5) via chromic acid, a strong oxidizer. In our experience, the published methods provided a yield between 10% and 20%. We speculated that the low yield might be due to formation of side products or poor extraction of the desired product. Consequently, instead of washing with brine and extracting with diethyl ether, we added 5% sodium hydrogen carbonate and extracted with dichloromethane while acidifying with hydrogen chloride. After purification via flash column chromatography, we achieved a typical yield of ~50%. Conversion of 1,1',2-tris-norsqualene aldehyde to 1,1',2-tris-norsqualenoyl acid using our updated methods has been repeated in house 3 times with yield ranging from 45% to 52% (Table 1). Chromic acid is not a suitable oxidizer for large scale production due to safety concerns. Silver(I) oxide and sodium chlorite are safer alternatives, but have lower resulting yields of 1,1',2-tris-norsqualenoyl acid (10% and 30% in house) [40, 41].
Yield comparison across synthesis methods to convert squalene into 1,1',2-tris-norsqualenoyl acid
| Maksimenko et al. [9] | In house replication | Methods reported herein | |
|---|---|---|---|
| 2-Bromo-3-hydroxysqualene (2) | 31% | 26% | 26% |
| 2,3-Oxidosqualene (3) | 98% | 97% | 97% |
| 1,1',2-Tris-norsqualene aldehyde (4) | 86% | 87% | 87% |
| 1,1',2-Tris-norsqualenoyl acid (5) | 35% | 18% | 52% |
In-house yields for the squalenoylation of gemcitabine using established procedures were approximately 15-20% [1]. To increase yield, we protected the two hydroxyl groups on gemcitabine with TBDMS-Cl using both DMAP and imidazole as catalysts. The resulting compound was confirmed with NMR (Figure S1) and mass spectrometry (Figure S2A). TBDMS-gemcitabine has increased solubility in dimethylformamide and reduced side reactions during squalenoylation compared to gemcitabine. TBDMS protection allowed us to selectively react gemcitabine with 1,1',2-tris-norsqualenoyl acid (5) by adapting the established procedures. We confirmed the intermediate product with mass spectrometry (Figure S2B) [1] without further column purification. To achieve the final product, we then removed the TBDMS groups (Scheme 1B) and obtained SqGem (8) at a 63% yield after purification (conjugation repeated 3 times, each starting with 0.5 g of SqAcid). SqGem was confirmed with mass spectrometry (Figure S2C) and NMR (Figure S3). A similar technique that used only DMAP as a catalyst has been previously reported but the yield was not described [42].
Benchtop NanoAssemblr allows for rapid nanoformation parameter exploration and reproducible mixing conditions
For NP formation, we used a scalable, microfluidic platform technology capable of precision mixing for the formation of NPs (Precision NanoSystems Benchtop NanoAssemblr). We wanted to evaluate nanoparticle size and PDI, aiming to minimize both quantities. Ideally, SqGem-based NPs will be less than 200 nm in diameter with a polydispersity index (PDI) less than 0.2. Two critical parameters that impact the microfluidic mixing were evaluated: the flow rate ratio (Figure 1A) and the total flow rate (Figure 1B). The flow rate ratio is the ratio of the aqueous solvent, 5% aqueous dextrose, to the organic solvent, acetone. Increasing the flow rate ratio creates smaller particles. A flow rate ratio that is too high, however, can disturb the precision mixing and increase PDI. Similarly, a high total flow rate can decrease nanoparticle size, but mixing suffers when the total flow rate surpasses a certain threshold. We found that a flow rate ratio of 4:1 minimized the particle size and PDI for a total flow rate of 14 mL/min. The resulting particle size and PDI were relatively insensitive to the total flow rate; however, a total flow rate of 14 mL/min reduced the variability in the mean size, as determined by the intensity. We found that a flow rate ratio of 4:1 and total flow rate of 14 mL/min yielded SqGem NPs of 70 ± 25 nm with a PDI of 0.18 and a zeta potential of approximately -10 mV (Figure 1A-B). All particles in this study were formed at ambient temperature and pressure.
Optimization of the particle assembly. (A) Effect of the flow rate ratio (aqueous solvent: organic solvent) on size and polydispersity index (PDI) at a constant flow rate of 12 mL/min. (B) Effect of the total flow rate on size and PDI. (C) Effect of SqGem:CholPEG:Chol on size and PDI at a flow rate ratio of 4:1 .
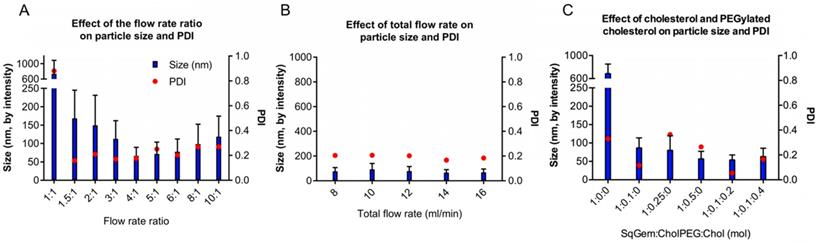
We also evaluated the inclusion of cholesterol-PEG-2k (CholPEG) (Figure 1C), which reportedly improves the stability of SqGem-based particles [43]. We did not observe a correlation between nanoparticle size and CholPEG concentration, as has been found using SqGem-based particles created in a drop-wise manner [1]. However, under the conditions considered here, the PDI was lower when we used a CholPEG concentration of 9 mol% (1:0.1:0 SqGem: CholPEG: Chol molar ratio) as compared with a CholPEG concentration of 0 mol%, 20 mol% or 33 mol% (1:0:0, 1:0.25:0 or 1:0.5:0 SqGem: CholPEG: Chol molar ratio, respectively). Thus, the SqGem-based particles we used in our in vitro studies were created with 9 mol% CholPEG, which yielded particles of 85 ± 30 nm with a PDI of 0.12. In our preferred solution (5% aqueous dextrose, w/v), CholPEG was necessary for the formation of SqGem-based NPs with microfluidics.
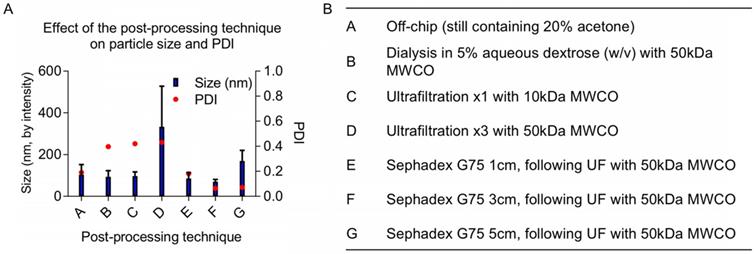
Once particles are assembled, either manually via dropwise techniques or using microfluidics, the organic solvent fraction must be removed before in vivo experiments. Established methods use rotary evaporation to pull off the organic fraction [1]. Although it is an effective way of removing solvents, rotary evaporation can be difficult to control because its efficiency depends on multiple parameters including pressure, temperature, rotation rate, initial total volume, aqueous to organic solvent ratio, and liquid surface area. We explored various post-processing techniques as an alternative: evaporation via increased air flow (data not shown), dialysis, size exclusion chromatography, and ultrafiltration. The increases in size and PDI following various post-processing techniques are likely due to NP fusion and aggregation when concentrated (using ultrafiltration) or incubated with acetone for multiple hours (using dialysis). Ultrafiltration is necessary to concentrate the off-chip particles before size-exclusion column purification; however, particles increase in diameter during ultrafiltration. We observed a minor batch-to-batch variability in final particle size using ultrafiltration, likely due to NP fusion with increasing concentration. Ultimately, we found that 50 kDa molecular weight cut-off (MWCO) ultrafiltration followed by purification with a Sephadex G-75 size exclusion column purification generated SqGem-CholPEGNPs between 70 and 130 nm with a PDI less than 0.25 (Figure 2). In addition to eliminating the acetone fraction (and other potential contaminants such as stray micelles), these post-processing steps narrowed the PDI. Once final post-processing methods were established, NPs were formulated for stability, microscopy, and in vitro studies at scales ranging from 0.5 to 4 mL (off-chip) with minor batch-to batch variability. For scale-up, microfluidics devices can be used in parallel to create NP batches at the liter-scale [44-47].
(A) Post-processing techniques alter final size and polydispersity index (PDI) of final NPs. (B) A variety of post-processing techniques were evaluated to remove the acetone from the SqGem-CholPEG NP solution and to ensure a narrow polydispersity index (PDI) of final NPs, including dialysis and combinations of ultrafiltration (UF) and size exclusion chromatography.
We also evaluated cholesterol inclusion at 15 mol% and 27 mol% (1:0.1:0.2 and 1:0.1:0.4 SqGem:CholPEG:Chol molar ratio, respectively) in order to increase stability in solution. Cholesterol inclusion did not alter the size of the resulting SqGem:CholPEG:Chol NPs, but cholesterol inclusion at 15 mol% was observed to yield a lower PDI (Figure 1C). We evaluated the effect of the SqGem:CholPEG:Chol ratio at final SqGem concentrations of 2 mg/mL and 4 mg/mL off-chip and confirmed that particles could form at higher concentrations without rapid aggregation.
Characterization of SqGem-CholPEG and SqGem-CholPEG-Chol NPs under likely storage conditions
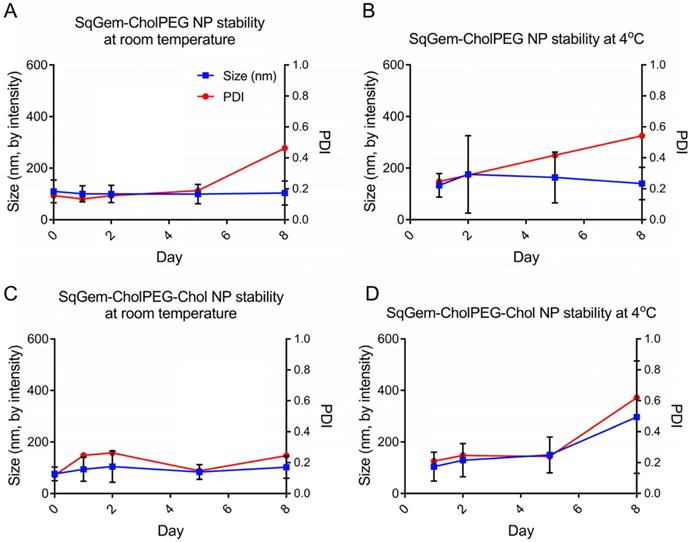
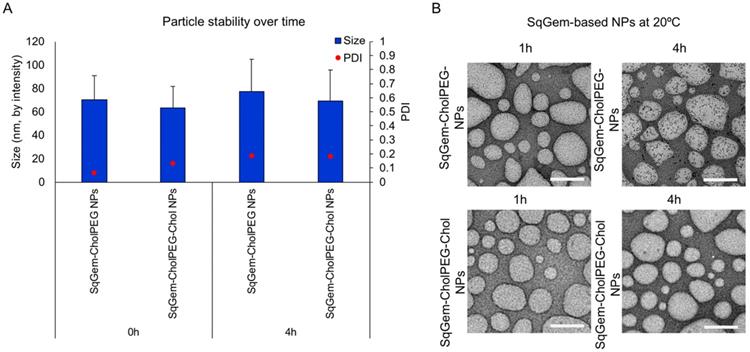
The particle size and PDI of SqGem-CholPEG NPs with and without 15 mol% Chol (1:0.1:0.2 SqGem:CholPEG:Chol molar ratio), was first assessed using DLS over 8 days of storage at either room temperature or 4°C. SqGem-CholPEG NPs maintained a diameter of 105 ± 35 nm and a PDI of approximately 0.16 for 5 days, after which the PDI increased (Figure 3A). When stored at 4°C, SqGem-CholPEG NPs increased in diameter to 130 ± 45 nm and their PDI increased to 0.25 within 24 hours (Figure 3B), and both measures continue to increase over subsequent days. The addition of 15 mol% cholesterol enhanced stability: SqGem-CholPEG-Chol NP diameter remained at 90 ± 40 nm with a PDI of approximately 0.2 throughout an 8-day incubation at room temperature (Figure 3C). When stored at 4°C, SqGem-CholPEG-Chol NPs remained stable in size and PDI for 5 days (Figure 3D).
Stability of SqGem-CholPEG and SqGem-CholPEG-Chol NPs over time and temperature. SqGem-CholPEG NPs stored at room temperature (A), SqGem-CholPEG NPs stored at 4°C (B), SqGem-CholPEG-Chol NPs stored at room temperature (C), and SqGem-CholPEG-Chol NPs stored at 4°C (D) were evaluated using dynamic light scatter to determine size and polydispersity index (PDI).
TEM and DLS were then applied to further characterize particle size and stability at room temperature (Figure 4) and 4°C (Figure S4). Variations between the sizes of nanoparticles measured using TEM and DLS result from differences in the sample preparation required by the techniques. TEM sample processing introduces aggregation of soft nanoparticles and requires dehydrating nanoparticles that typically exist in an aqueous medium [48]. Upon TEM, SqGem-CholPEG and SqGem-CholPEG-Chol NPs demonstrate a spherical shape, diameter on the order of 100-200 nm, and stability over 4 hours at room temperature (Figure 4A, B). At 4°C, the SqGem-CholPEG NPs assume a spindle-like morphology after 1h and increase in size compared to NPs containing Chol (Figure S4A, B). Although Chol prevents the formation of a spindle-like shape, morphological changes were still observed at 4°C with this formulation. Moreover, the morphological changes undergone during cooling are not reversible upon returning the particles to room temperature (Figure S5). The SqGem-based NPs described here are not stable when stored at 4°C. SqGem-CholPEG-Chol are stable for up to one week when stored at room temperature.
SqGem-CholPEG and SqGem-CholPEG-Chol NPs size and morphology. SqGem-CholPEG NPs with and without cholesterol (Chol) were evaluated at room temperature using dynamic light scatter to measure size and polydispersity index (PDI) (A) and transmission electron microscopy (B). Scale bar is equal to 200 nm.
SqGem-based NPs have similar or superior cytotoxicity profiles to free gemcitabine
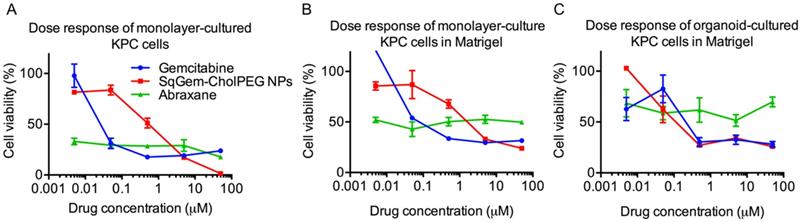
At low drug concentrations, we found that free gemcitabine was more toxic than SqGem-CholPEG NPs, as measured after 48h of incubation with monolayer-cultured KPC cells. The half maximal inhibitory concentration (IC50) values were accordingly lower for free gemcitabine than for SqGem-CholPEG NPs (Table 2). However, at higher concentrations, gemcitabine toxicity plateaus at ~15% cell viability, while viability approaches zero following SqGem-CholPEG NP incubation (Figure 5A). In KPC cells cultured as monolayers but plated in 3D Matrigel domes, viability following incubation with gemcitabine plateaus at 30%, with SqGem-CholPEG NPs showing similar cytotoxicity to free gemcitabine at the highest concentration tested (Figure 5B). Similarly, in KPC organoid culture, cell viability plateaus at 30% following incubation with either gemcitabine or SqGem-CholPEG NPs (Figure 5C). The cytotoxicity of SqGem-CholPEG NPs may be reduced in organoids either because subsets of cells within the organoid are less susceptible to SqGem or because the particles fail to completely penetrate the 3D organoids. Gemcitabine is similarly less effective against cells plated in organoid fashion compared to cells in monolayer culture. For comparison, we found that Abraxane was similarly cytotoxic across all drug concentrations tested (that is, did not display a dose-response curve similar to those of free gemcitabine or SqGem-CholPEG NPs), but exhibited less overall effectiveness against organoids and monolayer-cultured cells plated in Matrigel compared to monolayer-plated cells. IC50s could not be calculated for Abraxane using these experimental conditions and dosage range.
IC50 values across cell-culture techniques and drug treatments
| Treatment | |||
|---|---|---|---|
| Culture/plating technique | Gemcitabine | SqGem-CholPEG NPs | Abraxane |
| Monolayer/monolayer | 0.026 ± 0.01 µM | 0.530 ± 0.13 µM | NA**, <50% cell death |
| Monolayer/organoid | 0.023 ± 0.00 µM | 1.62 ± 0.15 µM | NA**, ~50% cell death |
| Organoid/organoid | 0.210 ± 0.07 µM* | 0.121 ± 0.04 µM* | NA**, <50% cell death |
* p < 0.05, Welch's t-test between Gemcitabine and SqGem-CholPEG NPs
** IC50 exceeded tested dosage
Efficacy of SqGem-CholPEG NPs compared to gemcitabine and Abraxane. Dose response curves of SqGem-CholPEG NPs, gemcitabine, and Abraxane in monolayer cultured KPC cells (A), monolayer-cultured cells plated in Matrigel in 3D (B), and in organoids plated in Matrigel (C).
Similar results were obtained for the cytotoxicity of SqGem-CholPEG NPs after only 24h of continuous drug incubation. At higher drug concentrations, cell viability neared 0% for the monolayer-cultured KPC cells incubated with SqGem-CholPEG NPs compared to 50% for free gemcitabine (Figure S6).
The cytotoxicity of SqGem-CholPEG-Chol NPs was also compared as a function of storage temperature in both monolayer-cultured PANC-1 (Figure 6A) and KPC cells (Figure 6B). No substantial effect of storage temperature was observed: NPs stored at both room temperature and at 4°C storage temperature caused a similar reduction in viability with increasing drug concentration. SqGem-CholPEG-Chol NPs again demonstrated significantly greater cytotoxicity than free gemcitabine at the higher drug concentrations tested, especially in the more drug-resistant human PANC-1 cell line.
Effect of temperature on efficacy and tolerability of squalene. Dose response curves of SqGem-CholPEG-Chol NPs, stored at room temperature and 4°C, and gemcitabine in monolayer-cultured PANC-1 cells (A) and KPC cells (B). Cell viability of PANC-1 and KPC cells after 48h incubation with squalene-only NPs at 20 µM (C).
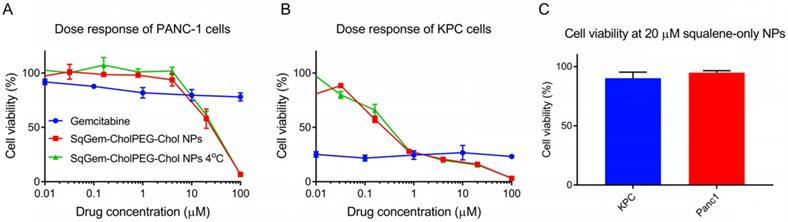
NPs containing squalene alone were also formed using the microfluidic methods, with a diameter of 700 ± 170 nm and a PDI of 0.33. Viability of PANC-1 cells and KPC cells was not reduced by incubation with squalene NPs up to a concentration of 20 µM, therefore we conclude that squalene does not contribute to the toxicity of SqGem-CholPEG NPs (Figure 6C). At higher concentrations, the squalene NPs failed to remain in suspension. These results are in agreement with reports on the safety and tolerability of squalene [49].
Squalenoylation of NOTA enables in vivo monitoring of SqGem NP pharmacokinetics and biodistribution
We modified squalene with an NHS ester to form an intermediate for further modification of squalene. The chemical structure and purity of SqNHS was confirmed with NMR (Figure S7), MALDI (Figure S8), and HPLC. Using this intermediate, we then squalenoylated NOTA, a 64Cu chelator (Scheme 2) and confirmed SqNOTA's chemical structure and purity using MALDI (Figure S9) and HPLC.
Synthesis of SqNOTA (10) from 1,1',2-Tris-norsqualenoyl acid (5) using SqNHS (9) as an intermediate.
After fabricating SqNOTA-SqGem-CholPEG-Chol NPs using a microfluidic platform, 64Cu was loaded onto the NPs or free SqNOTA. 64Cu loading efficiency was near 100% onto free SqNOTA and was 5% onto preformed SqNOTA-SqGem-CholPEG-Chol NPs. The difference in loading efficiency likely results from copper's limited access to NOTA within a PEGylated NP. Even with low 64Cu loading efficiency, 64Cu-SqNOTA-SqGem NPs, henceforth referred to as SqNOTA-SqGem NPs, were detectable over the 4h pharmacokinetic and organ distribution studies.
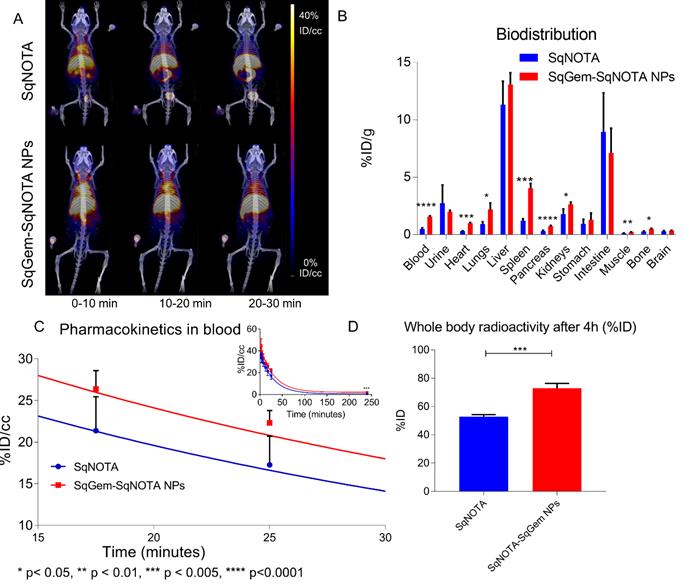
The pharmacokinetics of both SqNOTA and SqNOTA-SqGem NPs was monitored via PET imaging over 4 h after i.v. administration of a single dose. SqNOTA-SqGem NPs exhibit slower blood clearance than free SqNOTA. Throughout the first 30 minutes of circulation, the concentration of SqNOTA-SqGem NPs in blood, was greater than that of free SqNOTA, as visualized in the hearts of representative mice in PET/CT images (Figure 7A). After 4 h, the concentration of SqNOTA-SqGem NPs in the blood was 1.84% ID/cc, 3-fold higher than free SqNOTA at 0.57 % ID/cc (p = 0.0006). SqNOTA-SqGem NPs also remain in the heart, lungs, and spleen at significantly higher levels than free SqNOTA after 4 h (Figure 7B, Table S1). Based on the 4 hours of imaging data, the area under the curve for SqNOTA-SqGem NPs was greater than that for free SqNOTA (3,258 vs 2,472 %ID∙minutes/cc) (Figure 7C, Table S2). Additionally, whole body radioactivity of SqGem-SqNOTA NPs 4 h after injection (77.52 ± 3.44 %ID) was greater than that of SqNOTA at the same time point (52.84 ± 1.42 %ID) (Figure 7D). SqNOTA-SqGem NPs and free SqNOTA have similar blood half-lives, 21.36 and 20.24 minutes, respectively, based upon a single-phase decay. These data are in agreement with the reported plasma concentration of non-PEGylated SqGem NPs at similar time points when evaluated using liquid chromatography-tandem mass spectrometry [4]. Due to the low level of radioactivity remaining after 4 h of circulation, we were unable to collect longer timepoints to fit a two-phase decay model, which could better describe the pharmacokinetics of SqNOTA-SqGem NPs in terms of both an alpha and beta phase and provide a more accurate half-life.
Pharmacokinetics and biodistribution of SqNOTA and SqNOTA-SqGem NPs. Representative images of co-registered computed tomography-positron emission tomography of in vivo distribution of SqNOTA (top) and SqNOTA-SqGem NPS (bottom) showing percent injected dose per gram (%ID/g) for the first 30 minutes of circulation (A). Organ distribution of SqNOTA and SqNOTA-SqGem NPs expressed in percent injected dose per gram (%ID/g) (B). Time activity curve of SqNOTA and SqNOTA-SqGem NPs in blood expressed in percent injected dose per cubic centimeter (%ID/cc) (C). Whole body activity of SqNOTA and SqNOTA-SqGem NPs after 4 h (D).
Discussion
Squalenoylation has been proposed as a novel, promising strategy to enhance the efficacy of a host of therapeutics [1, 2, 5, 8, 9, 11, 12, 14, 50]. Because of the promise SqGem-based NPs hold for the treatment of pancreatic cancer in particular, we refined SqGem-based NP fabrication, evaluated the resulting in vitro activity in pancreatic cancer, and created a multifunctional NP to evaluate the pharmacokinetics and biodistribution of the NPs in vivo.
We adapted previously-reported methods to produce 1,1',2-trisnorsqualenoyl acid to increase chemical yield of the starting product for squalenoylation. In particular, we demonstrated the utility of protecting gemcitabine with TBDMS in the squalenoylation process. The resulting technique facilitated high yields of squalenoylated gemcitabine (here 63%) and could also be applied to enhance the squalenoylation yield of other bioactive compounds.
We also optimized microfluidic methods for the formation of SqGem-based NPs. SqGem-based NPs can self-assemble via nanoprecipitation, which is a “deceptively simple” process [51] relying on many parameters [52]. With the many variables of stir rate, surface area, infusion rate, solvent ratios, and particle concentration, the generation of monodisperse SqGem-based NPs using manual drop-wise techniques is challenging. By utilizing microfluidics to more precisely guide nanoprecipitation, we controlled or removed many of the variables involved in manual nanoprecipitation, providing reliable and repeatable nanoassembly. Importantly, the microfluidic device employed here is scalable, enabling eventual clinical translation of squalenoylated NPs [45].
The microfluidic platform also facilitated exploration of the impact of the cholesterol and Chol-PEG content on the formation, size, and stability of the SqGem-based NPs. We found that addition of CholPEG was required for SqGem-based NPs to form in 5% aqueous dextrose (w/v), but that large amounts of Chol-PEG lead to a wider distribution of particle sizes. The addition of Chol stabilized the size and PDI of SqGem-CholPEG-Chol NPs stored at room temperature but did not protect the SqGem-CholPEG-Chol NPs from morphological changes upon storage at 4°C. The size and PDI of SqGem-CholPEG and SqGem-CholPEG-Chol NPs were stable when stored at room temperature for 5 and 8 days, respectively. Although SqGem-CholPEG-Chol NPs changed morphology at a cooler temperature, their in vitro cytotoxicity was unaffected. Additional work is needed to further increase of SqGem-based particle stability. Freeze-drying SqGem-based NPs into a powder form, for example, has been shown to be a feasible method to prolong shelf life [53]. Combining the reliability of the microfluidic platform for nanoassembly with a freeze-dryer friendly formulation of SqGem-based NPs could greatly aid in clinical translation. Microfluidic systems provide flexibility and ease-of-use that may support a freeze-drying formulation.
In addition to evaluating and optimizing SqGem-based NP fabrication methods and formulation, we assessed the in vitro performance of SqGem-CholPEG and SqGem-CholPEG-Chol NPs in human and mouse lines of pancreatic cancer, including 3D organoids derived from the KPC genetically engineered mouse model of pancreatic cancer. Against monolayer-cultured pancreatic cancer cells, SqGem-CholPEG NPs were nearly 100% cytotoxic, whereas cell viability plateaued at high concentrations of gemcitabine and Abraxane.
Free gemcitabine has a short half-life, ranging from 32 to 94 minutes for short infusions in humans [54] and reported to be ~16 minutes in mice [55]. The clinical efficacy of gemcitabine is low due to a combination of this short plasma half-life and chemoresistance of pancreatic cancer cells, in addition to the challenging microenvironment and accelerated growth of pancreatic cancer. As shown previously using liquid chromatography-tandem mass spectrometry, squalenoylation of gemcitabine can increase its half-life 3.9-fold (based on two-exponential decay) [4]. Moreover, FRET imaging of SqGem-based NPs demonstrated that particles remain intact for up to 2h in circulation before destabilization [27]. Using a quantitative image-based method with multifunctional 64Cu-SqNOTA-SqGem NPs, we confirmed ~18% ID/cc of SqGem-based NP circulating after 30 minutes in mice, but the 4 h time course was insufficient to confirm the second phase of decay. Based upon SqGem-based NPs high uptake in the liver, lung and spleen, we also confirm clearance of these particles via the reticuloendothelial system.
We expect the dense stromal microenvironment of pancreatic cancer to remain a challenge for the delivery of SqGem-based NPs. In pancreatic cancer organoids, a more challenging in vitro model of pancreatic cancer, SqGem-CholPEG NPs did not fully eliminate viable tumor cells. We do not know whether this was due to the existence of a nonresponsive cell type within the heterogeneous cell population in the organoids, or because the nanoparticles particles fail to completely penetrate the 3D organoids.
In the future, techniques to target SqGem-based NPs to pancreatic cancer [8] and/or to alter the pancreatic cancer microenvironment to enhance SqGem-based NPs delivery will likely be necessary to enhance the efficacy of SqGem-based NPs in in vivo models of pancreatic cancer due to the dense, hypovascular stroma. Visualization of SqGem-based NPs in vivo using 64Cu-SqNOTA will facilitate future studies of interventional techniques such as high intensity focused ultrasound, which aim to increase NP deposition in primary tumors [33]. Additionally, 64Cu-SqNOTA can be applied for in vivo monitoring of other squalene-based NPs, allowing for assessment of biodistribution, accumulation, and pharmacokinetics [37].
Conclusions
Given its lethality, improved therapeutics for pancreatic cancer are desperately needed in the clinic. SqGem-based NPs show significant advantages over free gemcitabine in preclinical studies: specifically, SqGem-based NPs can overcome obstacles of drug resistance and the short plasma half-life of gemcitabine. To make a clinical impact, however, these NPs must be manufactured at large scale, display effectiveness in models of pancreatic cancer that mimic human pancreatic cancer and demonstrate in vivo reliability. Here, we present robust methods that build upon previous work to facilitate clinical translation of squalenoylated gemcitabine. In this study, we evaluate SqGem-based NP synthesis using techniques capable of scale-up to clinical production but performed at the 4 mL scale. We demonstrate that high concentrations of SqGem are effective against several in vitro pancreatic cancer cell lines, confirm short-term pharmacokinetic behavior of SqGem-based NPs, and present 64Cu-SqNOTA-SqGem NPs as effective multifunctional particles.
Materials and Methods (Additional chemical and pharmacokinetic characterization contained in the Supplemental Materials online)
Materials
Gemcitabine hydrochloride was purchased from LC Laboratories (Woburn, MA). Methoxy-PEG2k-cholesterol (CholPEG) was purchased from Nano CS (Boston, MA). Squalene (1, Scheme 1A) was used to produce 1,1',2-tris-norsqualenoyl aldehyde (4, Scheme 1A) by minimally adapting reported methods [9]. Organic compounds were purified using the Biotage Isolera Prime Flash Purification System (Charlotte, NC) and verified with NMR (Bruker Avance III) and mass spectrometry (Thermo Electron LTQ-Orbitrap XL Hybrid mass spectrometer). The NanoAssemblr Benchtop and microfluidic cartridges were purchased from Precision NanoSystems (Vancouver, Canada). All aqueous solvents used in nanoassembly were filtered through a 0.22 µm membrane before use. Other solvents and reagents were obtained from Sigma Aldrich (St. Louis, MO). Abraxane was a generous gift from Dr. Edward Kim (UC Davis Comprehensive Cancer Center). PANC-1 cells were purchased from American Type Culture Collection (CRL-1469, ATCC, Manassas, VA). KPC monolayer cells and organoids were a generous gift from Dr. David Tuveson (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). PANC-1 and KPC monolayer cells were cultured in Dulbecco's Modified Eagle medium (DMEM) containing 4500 mg/L glucose, L-glutamine and sodium pyruvate (# 11995, Invitrogen) and supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Organoids were cultured in Matrigel (BD Biosciences) domes (50 µL) in 24-well tissue-culture treated plates, bathed in 500 µL complete DMEM-F12 Advanced medium (supplemented with HEPES [1x, Invitrogen], GlutaMax [1x, Invitrogen], penicillin/streptomycin [1x, Invitrogen], B27 [1x, Invitrogen], N-acetyl-L-cysteine [1.25 mM, Sigma], recombinant human R-Spondin I [1 µg/mL, Peprotech], mNoggin recombinant protein [0.1 µg/mL, Peprotech], mouse epidermal growth factor [mEGF, 50 ng/mL, Invitrogen], Gastrin I [10 nM, Sigma], fibroblast growth factor 10 [FGF10, 100 ng/mL, Preprotech], Nicotinamide [10 mM, Sigma], A83-01 [0.5 µM, R&D Systems], and Rho Kinase Inhibitor [Y-27632, 10.5 µM, Sigma]). Organoids were cultured at 37ºC in a 5% CO2 atmosphere incubator. Medium was changed every 3-4 days and organoids were passaged into fresh Matrigel domes at least every 7 days.
All animal studies were approved by the University of California, Davis Animal Use and Care Committee. A total of 8 animals (female C57 Bl/6 mice, 8 weeks, 16-21 g) were studied.
Synthesis of 1,1',2-tris-norsqualenoyl acid (5)
Sulfuric acid (3.44 mL) was added to distilled water (35 mL) under stirring at 0°C. Then, sodium dichromate dihydrate (2.16 g) was slowly dissolved in the sulfuric acid solution at 0°C to obtain chromic acid. The chromic acid solution was added dropwise via syringe pump to a solution of 1,1',2-tris-norsqualene aldehyde (4, 2.7 g, 7.02 mmol) in diethyl ether (55 mL) at 0°C (all components were held at 0oC using ice packs). The reaction mixture was stirred at 0°C and the product formation was monitored by TLC (hexane/ethyl acetate, 3:1). After 2 hours, 1,1',2-tris-norsqualene aldehyde was not visible on TLC and 5% sodium hydrogen carbonate in water (50 mL) was added, the mixture was acidified to a pH of 2 with 10 M hydrochloric acid, and the crude organic product was extracted with dichloromethane (3 x 100 mL). The organic layers were dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The crude mixture was purified on a Biotage Isolera Prime™, a flash column chromatography system, using a SNAP KP-SIL cartridge (120 g, stepwise, 0-25%, 0.1% acetic acid in ethyl acetate to hexane, 1.5 column volume per 5% increase in ethyl acetate solution). 1,1',2-Tris-norsqualenoyl acid was obtained as a colorless oil (5, 1.46 g, yield = 52%). 1H-NMR (CDCl3, 800 MHz): δ(ppm) = 9.77 (t, J = 3.8 Hz, 1H); 5.19-1.10 (m, 5H); 2.53 (t, J = 15.1 Hz, 2H); 2.34 (t, J = 15.1 Hz, 2H); 2.14-1.98 (m, 16H); 1.70 (s, 3H); 1.64 (s, 3H); 1.61 (bs, 12H). ESI-MS m/z calculated for C27H44O2 [M + H]+ 401.34, found 401.34.
Conjugation of gemcitabine (6) to 1,1',2-tris-norsqualenoyl acid (5)
Protection of gemcitabine with TBDMS: Under nitrogen, gemcitabine hydrochloride (6, 0.50 g, 1.7 mmol) and imidazole (1.16 g, 17.0 mmol) were dissolved in anhydrous dimethylformamide and tert-butyldimethylsilyl chloride (TBDMS-Cl, 2.65 g, 10.0 mmol) was added. To the resulting solution, triethylamine (560 μL, 3.4 mmol) was added dropwise, followed by 4-dimethylaminophenol (DMAP). The reaction was monitored by TLC using ethyl acetate. After 48 hours, gemcitabine was no longer visible on TLC and the reaction was stopped and concentrated in vacuo to a viscous, yellow oil. Aqueous saturated sodium hydrogen carbonate (100 mL) was added and the product was extracted with ethyl acetate (4 x 100 mL). The combined organic extract was washed with saturated aqueous sodium hydrogen carbonate (1 x 50 mL), then saturated aqueous sodium chloride (1 x 50 mL). The organic layer was dried over anhydrous sodium sulfate. The organic solution was filtered, and the solvent was removed under reduced pressure to yield a pale yellow oil. The crude product was then dissolved in dichloromethane (200 mL) and washed with saturated aqueous sodium hydrogen carbonate (1 x 50 mL) and saturated aqueous sodium chloride (4 x 50 mL). The organic layer was dried over anhydrous sodium sulfate. The organic solution was filtered, and the solvent was removed under high vacuum to yield TBDMS-gemcitabine (7) as a waxy off-white solid without further purification (0.615 g). 1H-NMR (CDCl3, 800 MHz): δ(ppm) = 7.72 (d, J = 7.4 Hz, 1H); 6.36 (dd, J = 11.2 Hz, J = 3.8 Hz, 1H); 5.69 (d, J = 7.4 Hz, 1H); 4.33 (q, J = 10.5 Hz, 1H); 4.01 (d, J = 12.3 Hz, 1H); 3.92 (d, J = 8.4 Hz, 1H); 3.82 (d, J = 11.8 Hz, J = 2.0 Hz, 1H); 1.65 (bs, 4H); 0.95 (d, J = 26.3 Hz, 18H); 0.14 (t, J = 18.7 Hz, 12H). ESI-MS m/z calculated for C21H39F2N3O4Si2 [M + H]+ 492.24, found 492.25.
Preparation of 4-N-trisnorsqualenoyl-gemcitabine (8, SqGem): Under nitrogen, triethylamine (145 μL, 1.22 mmol) was added to a solution of 1,1',2-tris-norsqualenoyl acid (5, 0.43 g, 1.07 mmol) in anhydrous tetrahydrofuran (THF, 3 mL). The solution was cooled to -15ºC and a solution of ethyl chloroformate (ECF, 100 μL, 1.07 mmol) in anhydrous THF (3 mL) was added dropwise. The mixture was stirred at -15ºC for 30 minutes. Then, a solution of TBDMS-gemcitabine (0.5 g, 1.02 mmol) and triethylamine (145 μL, 1.22 mmol) in anhydrous THF (3 mL) was added to the reaction, dropwise. The reaction was allowed to return to room temperature. The reaction was monitored with TLC using hexane/ethyl acetate (3:1) and judged to be complete after 48 hours. The resulting solution was then concentrated in vacuo and the residue was taken up in aqueous sodium hydrogen carbonate (25 mL) and extracted with ethyl acetate (4 x 50 mL). The combined extracts were washed with sodium hydrogen carbonate (1 x 50 mL), then saturated aqueous sodium chloride (1 x 50 mL). The mixture was dried over anhydrous magnesium sulfate, filtered, and concentrated via rotary evaporation to yield the crude product as a colorless oil, which was used without purification for the deprotection step. ESI-MS m/z calculated for C48H81F2N3O5Si2 [M + H]+ 874.57, found 874.58.
The crude product was dissolved in anhydrous THF (10 mL) at 0oC under nitrogen. Tetra-n-butylammonium fluoride (TBAF, 1M) in THF (2.55 mL, 2.55 mmol) was added dropwise, under nitrogen, followed by acetic acid (145 μL, 2.55 mmol). The reaction was allowed to warm to room temperature. The reaction was monitored with TLC (hexane/ethyl acetate 3:1). After 24 hours, aqueous sodium carbonate (5 mL) was added and the reaction was left to stir for 30 minutes. The organic solvent was then removed via rotary evaporation. The remaining solution was taken up in aqueous sodium hydrogen carbonate (50 mL) and extracted with ethyl acetate (4 x 100 mL). The combined extracts were washed with 5% sodium hydrogen carbonate (50 mL), then saturated aqueous sodium chloride (50 mL). The mixture was dried over anhydrous magnesium sulfate, filtered, and concentrated via rotary evaporation to yield the crude product as a pale, yellow oil. The crude mixture was purified on a Biotage Isolera Prime purification system using a SNAP KP-SIL cartridge (100 g, stepwise, 0-5%, methanol in dichloromethane, 1.5 column volume per 1% increase in methanol) to yield 4-N-tris-norsqualenoyl-gemcitabine (8) as a white, amorphous solid (SqGem, 0.42 g , yield = 63% for the two steps).1H-NMR (CDCl3, 800 MHz): δ(ppm) = 7.98 (d, J = 7.3 Hz, 1H); 7.49 (d, J = 7.3 Hz, 1H); 6.27 (s, 1H); 5.24-5.09 (m, 5H); 4.58 (s, 1H); 4.11 (d, J = 12.3 Hz, 1H); 4.07 (dd, J = 8.2 Hz, J = 2.9 Hz, 1H); 3.98 (d, J = 12.5 Hz, 1H); 2.53 (t, J = 14.3 Hz, 2H); 2.38 (t, J = 13.5 Hz, 2H); 2.05 (m, 16H); 1.70 (s, 3H); 1.66 (s, 3H); 1.62 (m, 14H). ESI-MS m/z calculated for C36H53F2N3O5 [M + H]+ 646.40, found 646.41.
Nanoformation parameter exploration on the NanoAssemblr Benchtop
SqGem and methoxy-PEG2k-cholesterol (CholPEG) were dissolved in acetone at 10 mg/mL and 3.18 mg/mL, respectively. 5% dextrose in water (w/v) was passed through a 0.22 µM filter and used as the aqueous solvent. The total flow rate was held at 12 mL/min and the flow rate ratio (aqueous in mL/min: organic in mL/min) was varied from 1:1 to 10:1. After determining the optimal flow rate ratio, the total flow rate was varied from 10 to 16 mL/min while keeping the flow rate ratio at 4:1. The size of the particles in suspension off-chip was evaluated using a Malvern Zetasizer NS (Malvern Instruments Ltd., Malvern, UK). All particles throughout the study were formed at ambient temperature and pressure.
The Chol-PEG concentration was evaluated at a total flow rate of 12 mL/min and a flow rate ratio of 3:1. SqGem was dissolved in acetone at 8 mg/mL with a varying amount of CholPEG. 5% dextrose in water (w/v) was used as the aqueous solvent. The size of the particles in suspension off-chip was evaluated on a Malvern zetasizer NS. All nanoformation parameter exploration was performed using a 0.5 mL off-chip volume, at one run per trial, with a final SqGem concentration ranging from 3 mg/mL to 0.5 mg/mL. The selected parameters were then applied in subsequent studies.
Assembly of SqGem particles
Using the NanoAssemblr Benchtop (Precision NanoSystems), the SqGem acetone solution was streamed with a solution of sterile 5% aqueous dextrose (w/v) at a flow rate ratio of 4:1 (aqueous:organic) and a total flow rate of 14 mL/min. The initial 2 mL solution of NPs in 5% aqueous dextrose (w/v) and acetone was then concentrated via ultrafiltration using an Amicon Ultra 50 kDa centrifugal filter unit (Millipore Sigma) for 15 minutes at 15oC at 2000 x g to yield 500 μL of concentrated nanoparticles. The SqGem nanoassemblies were then separated from a small fraction of aggregate particles by passing the concentrated solution through a size exclusion gravity column of Sephadex G-75 (3 x 1 cm) equilibrated with 5% aqueous dextrose (w/v). The concentration of the final particles was evaluated using HPLC and the size and zeta potential were evaluated using a Malvern Zetasizer NS.
Stability studies
SqGem-CholPEG NPs and SqGem-CholPEG-Chol NPs were held at room temperature (20°C) and 4°C and imaged using transmission electron microscopy (TEM) at 1 and 4h post size-exclusion chromatography. At the same time points, the NPs were sized using dynamic light scattering (DLS). For TEM preparation, a 2.5 µl sample of freshly prepared SqGem-based NPs (1.0 mg/mL) was deposited on carbon-coated copper grids for 30 seconds before the excess solution was removed by blotting with filter paper. Subsequently, 2 µl of 2% uranyl acetate was applied to enhance the contrast for TEM imaging. The excess stain was blotted off and the samples were examined using a transmission electron microscope (JEOL, Peabody, MA, USA) at 100 kV and recorded by QTVIPS CCD camera (TVIPS, Gauting, Germany).
In vitro evaluation
The efficacy of SqGem-CholPEG NPs was first evaluated in vitro in KPC cells that were cultured as either monolayers or as 3D organoids. 2000 monolayer-cultured KPC cells were either plated as a monolayer or dispersed in Matrigel mounds (10 µL) to mimic the culturing conditions of an organoid in 96-well tissue culture treated plates. Organoid-cultured KPC cells (~2000 cells/well) were plated as organoids in Matrigel mounds (10 µL) in 96-well tissue culture treated plates. SqGem-CholPEG NPs with 9 mol% CholPEG were prepared using a drop-wise method as described in [1]. The SqGem-CholPEG NP solution was diluted in complete cell culture media to reduce the organic solvent fraction below 2%. Free gemcitabine, SqGem-CholPEG NPs, and Abraxane were incubated with the three variations of KPC cells for 48 hours (100 uL/well). Cell viability was evaluated via a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and compared to both no treatment control cells and cells incubated with 2% acetone.
SqGem-CholPEG-Chol and squalene NPs were fabricated on the NanoAssemblr and diluted in cell culture media to reduce the organic solvent fraction below 2%. SqGemCP-Chol NPs at room temperature, SqGem-CholPEG-Chol NPs at 4ºC, squalene NPs, and free gemcitabine were then incubated with monolayer-cultured and monolayer-plated KPC and PANC-1 cells for 48 hours before cell viability was assessed with an MTT assay and compared to no treatment control cells and 2% acetone-incubated cells.
Preparation of SqNOTA (10)
A solution of 1,1',2-tris-norsqualenoyl acid (5, 200 mg, 0.50 mmol) and N-hydroxysuccinimide (NHS, 57.5 mg, 0.50 mmol) in dichloromethane (5 mL) was made under nitrogen and 1-ethyl-3-(3-dimethlyaminopropyl)carbodiimide (EDAC, 77.5 mg, 0.50 mmol) was added. After 48h under magnetic stirring, the mixture was washed with water (50 mL) and extracted with dichloromethane (3 x 30 mL). The organic layers were dried over anhydrous magnesium sulfate and the solvent was removed via rotary evaporation. The resulting oil was purified via HPLC (30/70 acetonitrile/0.1% TFA in water to 5/95 acetonitrile/0.1% TFA in water over 30 mins at 60°C on C5) to yield 2,5-dioxopyrrolidin-1-yl (4E, 8E, 12E, 16E)- 4,8,13,17,21- petamethyldocosa- 4,8,12,16,20- pentaenoate (SqNHS, 9, 62 mg, 0.12 mmol, 24% yield). 1H-NMR (CDCl3, 800 MHz): δ(ppm) = 5.25-5.10 (m, 5H); 2.86 (bs, 4H); 2.73 (t, J = 15.1 Hz, 2H); 2.43 (t, J = 15.1 Hz, 2H); 2.14-1.97 (m, 16H); 1.70 (s, 3H); 1.66 (s, 3H); 1.62 (bs, 12H). SqNHS was verified with MALDI (Bruker UltraFlextreme MALDI TOF/TOF, Bruker Daltonics Inc., Billerica, MA) calculated for C31H47NO4 [M + Na]+ 520.34, found 520.48 and [M + K]+ 536.31, found 536.47.2-S-(4-aminobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA, 10 mg, 0.019 mmol) was dissolved in anhydrous acetonitrile (0.5 mL). SqNHS (15 mg, 0.029 mmol) in acetonitrile (0.5 mL) and triethylamine (27 µL, 0.193 mmol) were added to a solution at room temperature. After overnight reaction at shaker (1000 rpm), double distilled water (200 µL) was added, and solution kept shaking for additional one hour. Trifluoroacetic acid was added dropwise until the solution reached a pH of 2~3. The crude mixture was purified via HPLC (50/50 acetonitrile/0.05% TFA in water to 50/95 acetonitrile/0.05% TFA in water over 30 mins at 20°C on C5) to yield 2,2',2''-(2-(4-((4E,8E,12E,16E)-4,8,13,17,21-pentamethyldocosa-4,8,12,16,20-pentaenamido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (SqNOTA,10, 0.7 mg, 0.88 umol, 5% yield). SqNOTA was verified with MALDI (Bruker UltraFlextreme MALDI TOF/TOF, Bruker Daltonics Inc., Billerica, MA) calculated for C45H70N4O7 [M + H]+ 791.52, found 791.50.
Preparation of 64Cu-labeled SqNOTA and SqNOTA-SqGem NPs
Using the NanoAssemblr Benchtop (Precision NanoSystems), a solution of SqGem (10 mg/mL), Chol (1.2 mg/mL), Chol-PEG (3.1 mg/mL) and SqNOTA (0.159 mg/mL) in acetone was streamed with a solution of sterile 5% aqueous dextrose (w/v) at a flow rate ratio of 4:1 (aqueous:organic) and a total flow rate of 14 mL/min. The initial 1.6 mL solution of NPs in 5% aqueous dextrose (w/v) and acetone was then concentrated via ultrafiltration using an Amicon Ultra 50 kDa centrifugal filter unit (Millipore Sigma) for 45 minutes at 15°C at 2000 x g to yield 500 μL of concentrated nanoparticles. The SqNOTA-SqGem NPs were passed through a size exclusion gravity column of Sephadex G-75 (3 x 1 cm) equilibrated with PBS -/-.
The resulting solution of SqNOTA-SqGem NPs in PBS -/- (400 μL) was mixed with ammonium citrate (100 μL,0.1 M, pH 5.5) to adjust pH 5.5~6.0. 64CuCl2 (2 mCi, 2 μL) in 1M HCl was then added. The solution mixed for 90 minutes at 37°C before EDTA solution (0.1M, 5 μL) was added. 64Cu-SqNOTA-SqGem NPs was passed through a size exclusion gravity column of Sephadex G-75 (5 x 1 cm) equilibrated with 1xPBS -/- and 500 μL fractions were collected. The radioactivity of the resulting solution of 64Cu-SqNOTA-SqGem NPs was measured in a gamma counter.
SqNOTA (160 μg) was dissolved in ammonium citrate (20 μL, 0.1 M, pH 5.5). 64CuCl2 (2 mCi, 2 μL) in 1M HCl was then added. The solution mixed for 20 minutes at 20°C, and reaction completion was confirmed by instant radio thin layer chromatography. Free copper was removed using solid phase extraction with C18 cartridge. After the recovery 64Cu-labeled SqNOTA with 90% ethanol, evaporated, 64Cu-SqNOTA was reformulate in 1x PBS -/- and filtered through 0.2 um filter. Radiochemical purity was measured by ITLC and was higher than 99%.
PET-CT imaging and γ count
Briefly, two mice were placed side by side, anesthetized with isofluorane, and catheterized to ensure proper tail vein injection. Bolus injections of either SqNOTA or 64Cu-SqNOTA-SqGem NPs were administered as PET scans on a Inveon DPET scanner (Siemens) were initiated. Mice were imaged at 0 h and 4 h for 30 minutes on PET. Immediately following each PET scan, mice were imaged on a Siemens Inveon CT. Following the 4 h scans, mice were euthanized by injection of Euthasol and perfused with Dulbecco's Modification of Eagle's Medium (DMEM). Organs of interest were harvested and weighed, and radioactivity was measured by Wizard 1470 Automatic Gamma Counter (PerkinElmer), presented as a percent injected dose per gram organ (%ID/g). First 30 min PET images were segmented to 13 frames (15s4f, 30s2f, 60s3f, 300s3f, 600s1f) and reconstructed with maximum a posteriori algorithm. Blood radioactivity was calculated by drawing ROI of left ventricle of heart with PET/CT contrast normalized to 0 and 25 injected dose per cubic centimeter (%ID/cc) by a single observer with Inveon Research Workplace software (Siemens Preclinical Solutions). Using the same software, time activity curves (TACs) were obtained with region-of-interest (ROI) analysis and expressed as %ID/cc.
Abbreviations
SqGem: squalenoylated gemcitabine; NPs: nanoparticles; TBDMS: tert-butyldimethylsilyl; PEG: polyethylene glycol; DLS: dynamic light scatter; KPC: KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-Cre; PET: positron emission tomography; NOTA: 2-S-(4-aminobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid; SqAcid: 1,1',2-tris-norsqualenoyl acid; NMR: nuclear magnetic resonance; PDI: polydispersity index; CholPEG: cholesterol-PEG-2k; Chol: cholesterol; MWCO: molecular weight cut-off; IC50: half-maximal inhibitory concentration; NHS: n-hydroxy succinimide; MALDI: matrix-assisted laser desorption/ionization; HPLC: higher performance liquid chromatography; i.v.: intravenous; CT: computed tomography; ID: injected dose; cc: cubic centimeter
Acknowledgements
We appreciate the support of R01CA210553 and T32GM099608. NMR spectra were collected thanks to the support of DBIO722538.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaele D. et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6:2544-8
2. Desmaele D, Gref R, Couvreur P. Squalenoylation: a generic platform for nanoparticular drug delivery. J Control Release. 2012;161:609-18
3. Bildstein L, Dubernet C, Marsaud V, Chacun H, Nicolas V, Gueutin C. et al. Transmembrane diffusion of gemcitabine by a nanoparticulate squalenoyl prodrug: an original drug delivery pathway. J Control Release. 2010;147:163-70
4. Reddy LH, Khoury H, Paci A, Deroussent A, Ferreira H, Dubernet C. et al. Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice. Drug Metab Dispos. 2008;36:1570-7
5. Reddy LH, Renoir JM, Marsaud V, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Anticancer efficacy of squalenoyl gemcitabine nanomedicine on 60 human tumor cell panel and on experimental tumor. Mol Pharm. 2009;6:1526-35
6. Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J Control Release. 2007;124:20-7
7. Allain V, Bourgaux C, Couvreur P. Self-assembled nucleolipids: from supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 2012;40:1891-903
8. Bui DT, Nicolas J, Maksimenko A, Desmaele D, Couvreur P. Multifunctional squalene-based prodrug nanoparticles for targeted cancer therapy. Chem Commun (Camb). 2014;50:5336-8
9. Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH. et al. A unique squalenoylated and nonpegylated doxorubicin nanomedicine with systemic long-circulating properties and anticancer activity. Proc Natl Acad Sci U S A. 2014;111:E217-26
10. Othman M, Desmaele D, Couvreur P, Vander Elst L, Laurent S, Muller RN. et al. Synthesis and physicochemical characterization of new squalenoyl amphiphilic gadolinium complexes as nanoparticle contrast agents. Org Biomol Chem. 2011;9:4367-86
11. Raouane M, Desmaele D, Gilbert-Sirieix M, Gueutin C, Zouhiri F, Bourgaux C. et al. Synthesis, characterization, and in vivo delivery of siRNA-squalene nanoparticles targeting fusion oncogene in papillary thyroid carcinoma. Journal of medicinal chemistry. 2011;54:4067-76
12. Semiramoth N, Di Meo C, Zouhiri F, Said-Hassane F, Valetti S, Gorges R. et al. Self-assembled squalenoylated penicillin bioconjugates: an original approach for the treatment of intracellular infections. ACS nano. 2012;6:3820-31
13. Couvreur P, Reddy LH, Mangenot S, Poupaert JH, Desmaele D, Lepetre-Mouelhi S. et al. Discovery of new hexagonal supramolecular nanostructures formed by squalenoylation of an anticancer nucleoside analogue. Small. 2008;4:247-53
14. Caron J, Reddy LH, Lepetre-Mouelhi S, Wack S, Clayette P, Rogez-Kreuz C. et al. Squalenoyl nucleoside monophosphate nanoassemblies: new prodrug strategy for the delivery of nucleotide analogues. Bioorg Med Chem Lett. 2010;20:2761-4
15. Maksimenko A, Caron J, Mougin J, Desmaele D, Couvreur P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int J Pharm. 2015;482:38-46
16. Valetti S, Maione F, Mura S, Stella B, Desmaele D, Noiray M. et al. Peptide-functionalized nanoparticles for selective targeting of pancreatic tumor. J Control Release. 2014;192:29-39
17. Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine. 2011;7:841-9
18. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17:V7-V12
19. Cavalcante LD, Monteiro G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8-16
20. Reddy JA, Bloomfield A, Taylor C, Hargett K, Nelson M, Leamon C. Antitumor efficacy of EC1456 in patient derived xenograft models of ovarian, endometrial, NSCLC and TNBC. Cancer Res. 2015:75
21. Caron J, Maksimenko A, Mougin J, Couvreur P, Desmaele D. Combined antitumoral therapy with nanoassemblies of bolaform polyisoprenoyl paclitaxel/gemcitabine prodrugs. Polym Chem-Uk. 2014;5:1662-73
22. Harrisson S, Nicolas J, Maksimenko A, Bui DT, Mougin J, Couvreur P. Nanoparticles with In Vivo Anticancer Activity from Polymer Prodrug Amphiphiles Prepared by Living Radical Polymerization. Angew Chem Int Edit. 2013;52:1678-82
23. Bui DT, Maksimenko A, Desmaele D, Harrisson S, Vauthier C, Couvreur P. et al. Polymer Prodrug Nanoparticles Based on Naturally Occurring Isoprenoid for Anticancer Therapy. Biomacromolecules. 2013;14:2837-47
24. Boj SF, Hwang CI, Baker LA, Chio IIC, Engle DD, Corbo V. et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell. 2015;160:324-38
25. Maksimenko A, Bui DT, Desmaele D, Couvreur P, Nicolas J. Significant Tumor Growth Inhibition from Naturally Occurring Lipid-Containing Polymer Prodrug Nanoparticles Obtained by the Drug-Initiated Method. Chem Mater. 2014;26:3606-9
26. Gravier J, Sancey L, Hirsjarvi S, Rustique E, Passirani C, Benoit JP. et al. FRET imaging approaches for in vitro and in vivo characterization of synthetic lipid nanoparticles. Mol Pharm. 2014;11:3133-44
27. Cayre F, Mura S, Andreiuk B, Sobot D, Gouazou S, Desmaele D. et al. In Vivo FRET Imaging to Predict the Risk Associated with Hepatic Accumulation of Squalene-Based Prodrug Nanoparticles. Adv Healthc Mater. 2018:7
28. Arias JL, Reddy LH, Othman M, Gillet B, Desmaele D, Zouhiri F. et al. Squalene based nanocomposites: a new platform for the design of multifunctional pharmaceutical theragnostics. ACS nano. 2011;5:1513-21
29. Seo JW, Zhang H, Kukis DL, Meares CF, Ferrara KW. A Novel Method to Label Preformed Liposomes with 64Cu Positron Emission Tomography (PET) Imaging. Bioconjugate Chem. 2008;19:2577-84
30. Seo JW, Zhang H, Kukis DL, Meares CF, Ferrara KW. A novel method to label preformed liposomes with 64Cu for positron emission tomography (PET) imaging. Bioconjug Chem. 2008;19:2577-84
31. Seo JW, Ang J, Mahakian LM, Tam S, Fite B, Ingham ES. et al. Self-assembled 20-nm (64)Cu-micelles enhance accumulation in rat glioblastoma. J Control Release. 2015;220:51-60
32. Watson KD, Lai CY, Qin S, Kruse DE, Lin YC, Seo JW. et al. Ultrasound increases nanoparticle delivery by reducing intratumoral pressure and increasing transport in epithelial and epithelial-mesenchymal transition tumors. Cancer Res. 2012;72:1485-93
33. Wong AW, Fite BZ, Liu Y, Kheirolomoom A, Seo JW, Watson KD. et al. Ultrasound ablation enhances drug accumulation and survival in mammary carcinoma models. J Clin Invest. 2016;126:99-111
34. Rygh CB, Qin S, Seo JW, Mahakian LM, Zhang H, Adamson R. et al. Longitudinal investigation of permeability and distribution of macromolecules in mouse malignant transformation using PET. Clin Cancer Res. 2011;17:550-9
35. Wong AW, Ormsby E, Zhang H, Seo JW, Mahakian LM, Caskey CF. et al. A comparison of image contrast with (64)Cu-labeled long circulating liposomes and (18)F-FDG in a murine model of mammary carcinoma. Am J Nucl Med Mol Imaging. 2013;3:32-43
36. Dong H, Dube N, Shu JY, Seo JW, Mahakian LM, Ferrara KW. et al. Long-circulating 15 nm micelles based on amphiphilic 3-helix peptide-PEG conjugates. ACS nano. 2012;6:5320-9
37. Chakravarty R, Hong H, Cai WB. Positron Emission Tomography Image-Guided Drug Delivery: Current Status and Future Perspectives. Mol Pharmaceut. 2014;11:3777-97
38. Vantamelen EE, Curphey TJ. The Selective Invitro Oxidation of the Terminal Double Bonds in Squalene. Tetrahedron Lett. 1962:121-4
39. Van Tamelen EE. Bioorganic Chemistry - Sterols and Acyclic Terpene Terminal Epoxides. Accounts Chem Res. 1968;1:111 -+
40. Sen SE, Prestwich GD. Trisnorsqualene Alcohol, a Potent Inhibitor of Vertebrate Squalene Epoxidase. J Am Chem Soc. 1989;111:1508-10
41. Goupy P, Reynaud E, Dangles O, Caris-Veyrat C. Antioxidant activity of (all-E)-lycopene and synthetic apo-lycopenoids in a chemical model of oxidative stress in the gastro-intestinal tract. New J Chem. 2012;36:575-87
42. Maksimenko A, Mougin J, Mura S, Sliwinski E, Lepeltier E, Bourgaux C. et al. Polyisoprenoyl gemcitabine conjugates self assemble as nanoparticles, useful for cancer therapy. Cancer Lett. 2013;334:346-53
43. Bekkara-Aounallah F, Gref R, Othman M, Reddy LH, Pili B, Allain V. et al. Novel PEGylated Nanoassemblies Made of Self-Assembled Squalenoyl Nucleoside Analogues. Adv Funct Mater. 2008;18:3715-25
44. Belliveau NM, Huft J, Lin PJC, Chen S, Leung AKK, Leaver TJ. et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol Ther-Nucl Acids. 2012:1
45. Gdowski A, Johnson K, Shah S, Gryczynski I, Vishwanatha J, Ranjan A. Optimization and scale up of microfluidic nanolipomer production method for preclinical and potential clinical trials. J Nanobiotechnol. 2018:16
46. Liu DF, Cito S, Zhang YZ, Wang CF, Sikanen TM, Santos HA. A Versatile and Robust Microfluidic Platform Toward High Throughput Synthesis of Homogeneous Nanoparticles with Tunable Properties. Adv Mater. 2015;27:2298-304
47. Zhigaltsev IV, Belliveau N, Hafez I, Leung AKK, Huft J, Hansen C. et al. Bottom-Up Design and Synthesis of Limit Size Lipid Nanoparticle Systems with Aqueous and Triglyceride Cores Using Millisecond Microfluidic Mixing. Langmuir. 2012;28:3633-40
48. Bhise NS, Shmueli RB, Gonzalez J, Green JJ. A Novel Assay for Quantifying the Number of Plasmids Encapsulated by Polymer Nanoparticles. Small. 2012;8:367-73
49. Reddy LH, Couvreur P. Squalene: A natural triterpene for use in disease management and therapy. Adv Drug Deliver Rev. 2009;61:1412-26
50. Maksimenko A, Alami M, Zouhiri F, Brion JD, Pruvost A, Mougin J. et al. Therapeutic modalities of squalenoyl nanocomposites in colon cancer: an ongoing search for improved efficacy. ACS nano. 2014;8:2018-32
51. Lepeltier E, Bourgaux C, Amenitsch H, Rosilio V, Lepetre-Mouelhi S, Zouhiri F. et al. Influence of the nanoprecipitation conditions on the supramolecular structure of squalenoyled nanoparticles. Eur J Pharm Biopharm. 2015;96:89-95
52. Lepeltier E, Bourgaux C, Couvreur P. Nanoprecipitation and the "Ouzo effect": Application to drug delivery devices. Adv Drug Deliv Rev. 2014;71:86-97
53. Bildstein L, Hillaireau H, Desmaele D, Lepetre-Mouelhi S, Dubernet C, Couvreur P. Freeze-drying of squalenoylated nucleoside analogue nanoparticles. Int J Pharm. 2009;381:140-5
54. Company ELa. Gemzar (Gemcitabine HCl) for injection [package insert]. Administration FaD, editor. Indianapolis, Indiana. 2005
55. Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metabolism and Disposition. 1992;20:849-55
Author contact
![]() Corresponding author: kwferraredu
Corresponding author: kwferraredu