- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental Section
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2018; 2(2):168-183. doi:10.7150/ntno.23125 This issue Cite
Research Paper
Functionalised Carbon Nanotubes Enhance Brain Delivery of Amyloid-Targeting Pittsburgh Compound B (PiB)-Derived Ligands
Pedro Miguel Costa1, Julie Tzu-Wen Wang1#, Jean-François Morfin2#, Tamanna Khanum1, Wan To1, Jane Sosabowski3, Eva Tóth2 ![]() , Khuloud T Al-Jamal1
, Khuloud T Al-Jamal1 ![]()
1. School of Cancer & Pharmaceutical Sciences, Faculty of Life Sciences & Medicine, King's College London, SE1 9NH, London, United Kingdom.
2. Centre de Biophysique Moléculaire, UPR 4301, CNRS, Université d'Orléans, Rue Charles Sadron CS 80054, 45071, Orléans Cedex 2, France.
3. Centre for Molecular Oncology, Bart's Cancer Institute, Queen Mary University of London, London, EC1M 6BQ, United Kingdom.
# Authors share equal contribution
Received 2017-10-12; Accepted 2017-12-21; Published 2018-2-15
Abstract

Alzheimer's disease (AD) is a neurodegenerative disorder characterised by brain accumulation of toxic protein aggregates, including extracellular amyloid beta (Aβ) plaques, inflammation, neuronal death and progressive cognitive dysfunction. Current diagnostic modalities, based on cognitive tests, fail to detect early AD onset, thus emphasising the need to develop improved methods for pre-symptomatic disease detection.
Building on the properties of the Pittsburgh Compound B (PiB), an Aβ-binding molecule suitable to use as positron emission tomography (PET) imaging agent, and aiming at using a more clinically available modality (like magnetic ressonance imaging, MRI), PiB derivatives have been conjugated to the macrocyclic chelator 1,4,7-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecane (DO3A) monoamide. However, these derivatives do not readily cross the highly selective blood-brain barrier (BBB). Taking advantage of the capacity of functionalised carbon nanotubes (f-CNTs) to cross biological barriers, including the BBB, this manuscript reports on the conjugation of two PiB derivative Gd3+ complexes - Gd(L2) and Gd(L3) - to multi-walled f-CNTs (f-MWNTs) and assessment of their in vivo biodistribution and brain uptake. It is shown that Gd(L2) and Gd(L3) can be efficiently loaded onto different f-MWNTs, with significant improvement in brain accumulation of the conjugates compared to the free metal complexes.
Overall, this study demonstrates that f-MWNTs have potential to be used as carriers in theranostic applications involving brain delivery of BBB impermeable compounds.
Keywords: Alzheimer's disease, Aβ plaques, Pittsburgh Compound B, carbon nanotubes (CNTs), brain delivery
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder affecting mostly the elderly population, characterised by brain accumulation of protein aggregates, including intracellular neurofibrillary tangles (formed of hyperphosphorylated tau protein) and extracellular senile plaques (formed of fibrillary amyloid beta (Aβ) peptides) [1]. Accumulation of misfolded protein, in association with deficient immune response (chronic activation and disturbance of microglial clearance functions) [2], is the main cause of neuronal death and associated cognitive dysfunction seen in AD patients [3]. Current diagnostic methods, based on cognitive tests, fail to detect early AD onset, while imaging modalities such as magnetic resonance imaging (MRI) can detect structural brain changes only at advanced stages of disease [4]. In recent years substantial resources have also been invested in the identification of biomarkers in cerebrospinal fluid (CSF), to identify a prodromal AD signature that could improve diagnosis [5]. There is therefore a clear need to develop non-invasive neuroimaging-based methods for precise and early detection of this disease.
Accurate in vivo imaging of Aβ plaque deposition could be a fundamental step towards effective early diagnosis of AD, as well as an important tool for monitoring of therapy efficacy and disease progression. Over the last decade, several small organic Aβ-targeting compounds have been identified, including benzothiazoles, stilbenes [6] and thioflavin T derivatives, such as the Pittsburgh Compound B (PiB) [7]. Despite good blood-brain barrier (BBB)-crossing ability, high in vivo binding affinity towards Aβ and suitable properties for usage in positron emission tomography (PET), their clinical application is limited by short radioisotope lifetime, use of ionising radiation and low spatial resolution. In contrast to nuclear imaging modalities, MRI allows non-invasive imaging of brain structures with superior spatial resolution, avoiding patient exposure to ionising radiation and at a smaller cost. MRI resolution can be further enhanced by paramagnetic contrast agents, including gadolinium (Gd3+) chelates or iron oxide nanoparticles [8]. Given the high electronic spin and slow relaxation times, Gd3+ complexes are the most widely used MRI contrast materials [8]. Selenium-based nanoparticles have also been successfully tested for Aβ binding and disruption, although their in vivo activity has yet to be demonstrated [9, 10].
Aiming at developing metal-based multimodal imaging probes for detection of Aβ plaques, PiB derivatives have been synthesised by conjugation to 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (DO3A) monoamide [11, 12], a non-ionic macrocyclic chelator that forms stable, neutral complexes with trivalent metal ions, including Gd3+ (for MRI) and Indium-111 (111In3+) (for single-photon emission computed tomography, SPECT). Moreover, the intrinsic fluorescence of the PiB moiety [13] could potentially allow for high-resolution optical microscopy tracing of the imaging probe. While ex vivo staining of human AD brain tissue showed that these derivatives selectively target Aβ plaques [11], their clinical application is limited by the reduced ability to cross the BBB [11, 12]. The BBB permeability of a compound is directly influenced by three main physico-chemical and physiological parameters: (i) lipophilicity, represented by the water/octanol partition coefficient (log Poct/water), (ii) molecular weight and (iii) plasma pharmacokinetics [14]. In previous studies, it was shown that the lipophilicity of the PiB derivatives Gd(L1) and Gd(L2) (log Poct/water = -0.15 and 0.03, respectively) [11, 12] is reduced compared to the highly lipophilic PiB (log Poct/water = 1.23) and other compounds with optimal BBB permeation (log Poct/water ~2) [15]. Biodistribution experiments in healthy adult Swiss mice using an 111In-labelled analogue of Gd(L1) have also revealed moderate BBB penetration (0.36 % of the injected dose per gram of tissue in the cortex at 2 min after injection) [11].
To improve brain delivery, impermeable compounds could be conjugated to nanocarriers capable of translocating across the BBB - either per se or upon targeting to transporters expressed in the brain vasculature - and reaching the desired regions in the brain [16]. In this regard, carbon nanotubes (CNTs) display unique physico-chemical properties suitable for use in nanomedicine applications, including large surface area (allowing conjugation of therapeutic and imaging molecules), electrochemical stability and a high aspect ratio [17-19]. Further surface modification/functionalisation with hydrophilic molecules, polymers or other biocompatible molecules can enhance CNT water dispersibility, reduce toxicity and improve target cell/tissue delivery [20-23]. Importantly, the ability of functionalised CNTs (f-CNTs) to cross biological barriers by energy-dependent (transcytosis) and independent (passive) mechanisms, as well as intrinsic optical and thermal properties, constitutes a major CNT advantage for brain-targeted theranostic applications, compared to other nanocarriers [24]. Previous in vitro and in vivo proof-of-concept studies from our group demonstrated that functionalised multi-walled carbon nanotubes (f-MWNTs) cross an intact BBB [25-27]. Functional degradation, including loss of cylindrical structure, was also reported for stereotactically-administered f-MWNTs in mouse brain, most probably mediated by peroxidases and other degradative enzymes released by microglia [28].
Aiming at improving brain delivery of optimised PiB derivatives, in this study two different complexes - Gd(L2) and Gd(L3) (Fig. 1) - were non-covalently conjugated to f-MWNTs. MWNT characterisation was performed using thermogravimetric analysis (TGA) and Kaiser test, while conjugation was evaluated using inductively coupled plasma mass spectrometry (ICP-MS) and fluorimetry. Finally, the ligands L2/L3 were conjugated to f-MWNTs and radiolabelled with 111In, thus allowing quantification of in vivo biodistribution and brain uptake by SPECT/CT and γ-scintigraphy.
Structure of ligands.
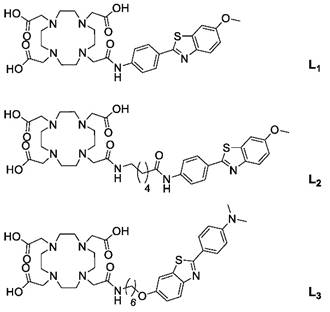
Experimental Section
Reagents and solutions
Pristine MWNTs (NC3100, Batch #120710, > 95% wt. purity, average dimensions of 1.5µm (length) and 9.5 nm (diameter)), were purchased from Nanocyl SA (Sambreville, Belgium). Chemicals (listed in Supplementary Materials) were purchased from Fisher Scientific, Sigma-Aldrich, Alfa Aesar, TCI Europe and CheMatech, and were used without further purification. Analytical grade solvents were used and were not purified further unless specified. Isopore hydrophilic and hydrophobic membrane filters (0.2 µm pore size) were purchased from Merck Millipore (Watford, UK). The radioactive probe 111InCl3 as an aqueous solution in 0.5 M HCl was purchased from Mallinckrodt Pharmaceuticals (The Netherlands) which is used directly without further purification. Instant thin layer chromatography paper impregnated with silica gel (iTLC-SG) was obtained from Agilent Technologies (UK). Fetal bovine serum was purchased from First Link Ltd (UK). All reagents and analytical grade solvents were used without further purification, unless specified. Other materials and equipment used for chemical synthesis are listed in Supplementary Material.
Synthesis of Gd(L2) and Gd(L3)
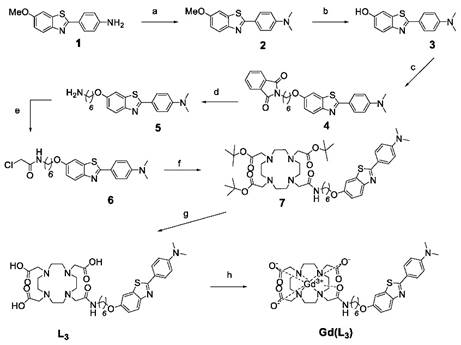
Gd(L3) was synthesised as shown in Scheme 1. The synthesis of Gd(L2) has been previously reported [12]. For the synthesis of Gd(L3), reactions were monitored by thin-layer chromatography (TLC) on Kieselgel 60 F254 (Merck) on an aluminium support, with detection by examination under UV light (254 nm), by spraying with ninhydrin, and by coloration of the complex with Dragendorff solutions. Polar affinity chromatography was performed with silica gel (SigmaAldrich). 1H and 13C NMR spectra were recorded on a Bruker Avance Spectrometer using a 5mm BBFO probe at 599.903 and 150.860 MHz for 1 H and 13C, respectively. High Resolution Mass spectrometry (HRMS) was performed at the Centre de Biophysique Moléculaire du CNRS in Orléans, France.
Synthesis of Gd(L3). The reagents and conditions were as follows: a) paraformaldehyde, NaCNBH3, AcOH, RT, 12h, 97%; b) BBr3, CH2Cl2, 0°C, 1h, 99%; c) N-(6-Bromohexyl)phthalimide, K2CO3, Acetone, reflux, 72h, 86%; d) NH2NH2, reflux, 2h, 99%; e) Chloroacetyl Chloride, NEt3, THF, 0°C, 3h, 83%; f) DO3A-tBu, K2CO3, CH3CN, reflux, 16h, 76%; g) TFA, CH2Cl2, RT, 3h, 82%; h) GdCl3, pH 5,5, 2h.
4-(6-methoxybenzo[d]thiazol-2-yl)-N,N-dimethylaniline: 2
In a round flask, 4-(6-methoxybenzo [d]thiazol-2-yl)aniline 1 (500mg, 1.95 mmol) and paraformaldehyde (600mg, 20 mmol) were dissolved in 50ml of acetic acid. After 1h at room temperature, sodium cyanoborohydride (400mg, 6.36 mmol) was added and the solution was mixed overnight. The reaction was neutralised by adding ammonium hydroxide. A yellowish precipitate was formed, collected and washed with water to give, without further purifications, 538mg (97%) of compound 2.
1H NMR (CDCl3, 298K, 599.902 MHz): 3.05 (s, 6H, N-(CH3)2); 3.88 (s, 3H, -OCH3); 6.74 (d, 3J= 8.9Hz, 2Har); 7.03 (dd, 3J= 8.9Hz, 4J= 2.5Hz, 1Har); 7.32 (d, 4J= 2.5Hz, 1Har); 7.86 (d, 3J= 8.9Hz, 1Har); 7.90 (d, 3J= 8.9Hz, 2Har)
13C NMR (CDCl3, 298 K, 150.86 MHz): 42.4 (2C, -N-(CH3)2); 56.0 (1C, -OCH3); 104.6 (1Car); 112.0 (2Car); 115.1 (1Car); 121.8 (1Car); 123.0 (1Car); 128.7 (2 Car); 136.0 (1Car); 152.1 (1Car); 157.3 (1Car); 166.7 (1Car)
2-(4-(dimethylamino)phenyl)benzo[d]thiazol-6-ol: 3
Compound 2 (500 mg, 1.76 mmol) was dissolved in 12ml of dichloromethane at 0°C and 4ml of boron tribromine 1M were added dropwise. The solution was mixed for 3h and neutralised by adding a solution of sodium hydroxide 1N and the extraction was made with ethyl acetate (3X 30ml). The organic fraction was collected, dried over magnesium sulfate and evaporated to give, without further purifications, 471mg (99%) of product 3.
1H NMR (DMSO-d6, 298K, 599.902 MHz): 3.07 (s, 6H, -N-(CH3)2); 6.86 (d, 3J= 9.0Hz, 2Har); 6.98 (dd, 3J= 8.7Hz, 4J= 2.4Hz, 1Har); 7.39 (d, 4J= 2.4Hz, 1Har); 7.78 (d, 3J= 8.7Hz, 1Har); 7.86 (d, 3J= 9.0Hz, 2Har), 9.71 (1H, OH)
2-(6-((2-(4-(dimethylamino)phenyl)benzo[d]thiazol-6-yl)oxy)hexyl)isoindoline-1,3-dione: 4
To a solution of N-(6-Bromohexyl)phthalimide (860mg, 2.77mmol) in 60ml of acetone were added 1g of potassium carbonate and a solution of compound 3 (450mg, 1,66mmol) in 10ml of acetone. The mixture was refluxed 3 days. After cooling down to room temperature, the solid was filtered off and the solvent was evaporated. Crude product was dissolved in 5ml of dichloromethane and petroleum ether was added until precipitation. The white solid was isolated to give 715mg (86%) of compound 4.
1H NMR (CDCl3, 298K, 599.902 MHz): 1.44 (m, 2H, -CH2-CH2-CH2-); 1.54 (m, 2H, -CH2-CH2-CH2-); 1.73 (m, 2H, -CH2-CH2-CH2-); 1.82 (m, 2H, -CH2-CH2-CH2-); 3.05 (s, 6H, -N-(CH3)2); 3.71 (t, 3J= 7.2Hz, 2H, -CH2-CH2-NPht); 4.00 (t, 3J= 6.4Hz, 2H, -CH2O-); 6.74 (d, 3J= 8.9Hz, 2Har); 7.03 (dd, 3J= 8.9Hz, 4J= 2.4Hz, 1Har); 7.28 (d, 4J= 2.4Hz, 1Har); 7.70 (m, 2HPht); 7.85(m, 1Har, 2HPht); 7,90 (d, 3J= 8.9Hz, 2Har).
13C NMR (CDCl3, 298 K, 150.86 MHz): 25.7(1C, -CH2-CH2-CH2-); 26.6 (1C, -CH2-CH2-CH2-); 28.5 (1C, -CH2-CH2-CH2-); 29.2 (1C, -CH2-CH2-CH2-); 37.9 (1C, -CH2-CH2-NPht); 40.1 (2C, -N-(CH3)2); 68.4 (1C, -OCH2-); 105.1 (1Car); 111.7 (2Car); 115.3 (1Car); 121.6 (1Car); 122.7 (1Car); 123.1 (2CPht); 128.5 (2Car); 132.1 (2CPht); 133.8 (2CPht); 135.8 (1Car); 148.8 (1Car); 151.8 (1Car); 156.5 (1Car); 166.2 (1Car); 168.4 (2C, COPht)
HRMS (ESI): m/z for C29H29N3O3S, calculated 499.1930; found 500.2005 [M+H]+
4-(6-((6-aminohexyl)oxy)benzo[d]thiazol-2-yl)-N,N-dimethylaniline: 5
Compound 4 (700mg, 1.40mmol) was refluxed 2h in 15 ml of hydrazine hydrate. After cooling down, product was extracted with dichloromethane (3 x 30ml) and the organic fraction was washed twice with 10 ml of brine. The organic phase was dried over magnesium sulphate and evaporated to afford to 512mg (99%) of compound 5 as a yellow oil.
1H NMR (CDCl3, 298K, 599.902 MHz): 1.42 (m, 4H, CH2-CH2-CH2); 1.48 (m, 2H, -CH2-CH2-CH2-); 1.83 (m, 2H, -CH2-CH2-CH2-); 2.72 (t, 3J= 6.4Hz, 2H, -CH2-CH2-NH2); 3.05 (s, 6H, -N-(CH3)2); 4.02 (t, 2H, 3J= 6.4Hz, -CH2O-); 6.74 (d, 3J= 8.9Hz, 2Har); 7.02 (dd, 3J= 8.9Hz, 4J= 2.4Hz, 1Har); 7.30 (d, 4J= 2.4Hz, 1Har); 7.84 (d, 1H, Har); 7,90 (d, 3J= 8.9Hz, 2Har)
13C NMR (CDCl3, 298 K, 150.860 MHz): 26.0 (1C, -CH2-CH2-CH2-); 26.7 (1C, -CH2-CH2-CH2-); 29.3 (1C, -CH2-CH2-CH2-); 33.8 (1C, -CH2-CH2-CH2-); 40.2 (2C, -N-(CH3)2); 42.2 (1C, -CH2-NH2); 68.5 (1C, -OCH2-); 105.1 (1Car); 111.7 (2Car); 115.4 (1Car); 121.7 (1Car); 122.7 (1Car); 128.5 (2Car); 135.8 (1Car); 148.9 (1Car); 151.9 (1Car); 156.6 (1Car); 166.4 (1Car)
HRMS (ESI): m/z for C21H27N3OS, calculated 369.1875; found 370.1946 [M+H]+ and 371.2032 [M+2H]2+
2-chloro-N-(6-((2-(4-(dimethylamino)phenyl)benzo[d]thiazol-6-yl)oxy)hexyl)acetamide: 6
To a solution of compound 5 (500mg, 1.35mmol) and triethylamine (565µl, 4.06mmol) in 15ml of dry THF at 0°C, was added dropwise a solution of chloroacetyl chloride (650µl, 4.06mmol) in 5 ml of dry THF. The mixture was stirred 3H and 60 ml of water was added. The solution was stored at 4°C overnight. Yellowish crystals were filtered off to give 499mg (83%) of compound 6.
1H NMR (CDCl3, 298K, 599.902 MHz): 1.42 (m, 4H, -CH2-CH2-CH2-); 1.48 (m, 2H, -CH2-CH2-CH2-); 1.83 (m, 2H, -CH2-CH2-CH2-); 3.05 (s, 6H, -N-(CH3)2); 3.35 (dt, 3J= 6.1Hz, 3J= 7.2Hz, 2H, -CH2-CH2-NHCO-); 4.02 (t, 3J= 6.4Hz, 2H, -CH2O-); 4.05 (s, 2H, -CH2-Cl); 6.59 (bs, 1H, -NHCO); 6.74 (d, 3J= 8.9Hz, 2Har); 7.02 (dd, 3J= 8.9Hz, 4J= 2.4Hz, 1Har); 7.30 (d, 4J= 2.4Hz, 1Har); 7.86 (d, 1H, Har); 7,90 (d, 3J= 8.9Hz, 2Har)
13C NMR (CDCl3, 298 K, 150.860 MHz): 25.8 (1C, -CH2-CH2-CH2-); 26.6 (1C, -CH2-CH2-CH2-); 29.2 (1C, -CH2-CH2-CH2-); 29.3 (1C, -CH2-CH2-CH2-); 39.8 (1C, -CH2-NH2); 40.2 (2C, -N-(CH3)2); 42.7 (1C, -CH2-Cl); 68.4 (1C, -OCH2-); 105.1 (1Car); 111.8 (2Car); 115.4 (1Car); 122.6 (1Car); 125.5 (1Car); 128.6 (2Car); 135.8 (1Car); 148.9 (1Car); 151.9 (1Car); 156.6(1Car); 165.8 (1CO); 166.4 (1Car)
HRMS (ESI): m/z for C23H28ClN3O2S, calculated 445.1591; found 446.1663 [M+H]+
2,2',2''-(10-(2-((6-((2-(4-(dimethylamino)phenyl)benzo[d]thiazol-6-yl)oxy)hexyl)amino)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid: 7
To a solution of DO3A-tBu (300mg, 0.58mmol) and potassium carbonate (226 mg, 2 mmol) in 20 mL of dry acetonitrile was added a solution of 6 (310mg, 0.7 mmol) in 15 mL of dry acetonitrile. The reaction mixture was stirred for 16 h at 90°C. After cooling, the solid was filtered off and the solvent was evaporated. The crude product was purified by flash chromatography (eluent: ethyl acetate/methanol 95/5) and 407mg (76%) of product 7 were isolated as a yellow oil.
1H NMR (CDCl3, 298K, 599.902 MHz): 1.40 (m, 2H, -CH2-CH2-CH2-); 1.44 (s, 18H, -C(CH3)3); 1.45 (s, 9H, -C(CH3)3); 1.48 (m, 2H, -CH2-CH2-CH2-); 1.61 (m, 2H, -CH2-CH2-CH2-); 1.81 (m, 2H, -CH2-CH2-CH2-); 2.25 (bs, 8H, -NCH2-CH2N-); 2.55 (bs, 4H, -NCH2-CH2N-); 2.85 (bs, 6H, -NCH2-CH2N-, -N-CH2-CONH-); 3.05 (s, 6H, -N-(CH3)2); 3.22 (dt, 3J= 6.1Hz, 3J= 5.9Hz, 2H, -CH2-NHCO-); 3.39 (bs, 6H, -N-CH2-COOtBu); 3.99 (t, 3J= 6.4Hz, 2H, -CH2O-); 6.74 (d, 3J= 8.9Hz, 2Har); 7.0 (dd, 3J= 8.9Hz, 4J= 2.4Hz, 1Har); 7.31 (d, 4J= 2.4Hz, 1Har); 7.62 (t, 1H, NHCO); 7.84 (d, 3J= 8.9Hz, Har); 7,90 (d, 2Har)
13C NMR (CDCl3, 298 K, 150.860 MHz): 25.4 (1C, -CH2-CH2-CH2-); 26.4 (1C, -CH2-CH2-CH2-); 27.5 (3C, -C(CH3)3); 27.6 (6C, -C(CH3)3) ;28.8 (1C, -CH2-CH2-CH2-); 29.1 (1C, -CH2-CH2-CH2-); 38.7 (1C, -CH2-NHCO-); 39.9 (2C, -N-(CH3)2); 48.1 (4C, -NCH2-CH2N-); 52.0 (4C, -NCH2-CH2N-); 55.1 (2C, -N-CH2-COOtBu); 55.3 (1C, -N-CH2-COOtBu); 56.0 (1C, -N-CH2-CONH-); 68.2 (1C, -OCH2-); 81.2 (2C, -C(CH3)3); 81.3 (1C, -C(CH3)3); 104.7 (1Car); 111.3 (2Car); 115.1 (1Car); 120.9 (1Car); 122.1 (1Car); 128.1 (2Car); 135.3 (1Car); 148.3 (1Car); 151.5 (1Car); 156.3 (1Car); 165.9 (1Car); 171.0 (1C, -CONH-); 171.8 (2C, -COOtBu); 172.1 (1C, -COOtBu)
HRMS (ESI): m/z for C49H77N7O8S calculated 923.5554; found 924.5622 [M+H]+
tri-tert-butyl 2,2',2''-(10-(2-((6-((2-(4-(dimethylamino)phenyl)benzo[d]thiazol-6-yl)oxy)hexyl)amino)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate: L3
To a solution of 7 (350mg, 0.38mmol) in 10 ml of dichloromethane was added 10ml of trifluoroacetic acid. The solution was stirred 3h at room temperature and the solvents were evaporated. The crude product was purified by flash chromatography on RPC18 silica (eluent: H20/Methanol). 207mg (82%) of ligand L3 were isolated as a yellow powder.
1H NMR (CDCl3, 298K, 599.902 MHz): 1.03 (bs, 4H, -CH2-CH2-CH2-); 1.28 (bs, 4H, -CH2-CH2-CH2-); 2.28 (bs, 6H, NCH2-CH2N, CH2-NHCO); 3.10-3.26 (bs, 20H, NCH2-CH2N, CH2-NHCO, N-(CH3)2); 3.57 (bs, 4H, N-CH2-COOH); 3.74 (bs, 4H, N-CH2-COOH, CH2O); 5.66 (bs, 2Har); 6.06 (bs, 1Har); 6.51 (bs, 1Har); 6.95 (bs, 1H, Har); 7.33 (d, 1H, Har)
HRMS (ESI): m/z for C37H53N7O8S calculated 755.3676; found 757.3824 [M+2H]2+ and 758.3910 [M+3H]3+
General procedure for the preparation of gadolinium (III) complexes
Gd(Lx) (x = 2,3) complexes were prepared by mixing aqueous solutions of GdCl3 and the ligand Lx in equimolar quantities and adjusting the pH to 5.5 with aqueous NaOH (0.1 mM). The solutions were allowed to react for 2h at room temperature while the pH was continuously adjusted. The absence of free metal ion was checked in each sample using the xylenol orange test.
Determination of Octanol-Water Partition Coefficient (log Poct/water)
The partition coefficient of Gd(L3) was determined, following the method previously used, as the ratio of the concentration of the compound in octanol and concentration of the compound in the aqueous phase [11, 12].
The logarithm of the partition coefficient is referred to as the log P value. The ''shake flask'' method was used for the determination of log P. Water saturated with octanol and octanol saturated with water were used in the experiments. The benzothiazol ring absorbs strongly at approximately 330 nm; therefore, the partition was quantified using UV spectrophotometry with a PerkinElmer Lambda 19 UV-vis spectrophotometer. For each phase, the maximum wavelength was verified. A 1:1 volume ratio was used for the partitioning of the solution with Gd(L3). In a 2-mL Eppendorf tube, 0.5 mL of a 100 µM solution of Gd(L3) was added to 0.5 mL of the saturated phase of 1-octanol. Each sample was centrifuged for 30 min. Gd(L3) concentrations were determined in each phase using standard curves.
Synthesis of functionalised MWNTs (f-MWNTs)
The synthesis of f-MWNTs was performed in three sequential steps as described in Scheme 2 and previously reported [29].
Synthesis of functionalised MWNTs (f-MWNTs). Pristine MWNTs were incubated under strong acidic conditions to produce oxidised MWNTs (ox-MWNTs). The carboxylic acid moieties of ox-MWNTs were reacted with a phthalimide-protected amine-terminated spacer via amide coupling reaction, in the presence of N-hydroxybenzotriazole (HOBt) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (1:2:2:2 molar ratio). Cleavage of the protection group was performed under strong alkaline conditions using hydrazine, yielding carboxylate and amine-functionalised MWNTs (ox-MWNTs-NH3+). RT: room temperature; DMF: dimethylformamide.
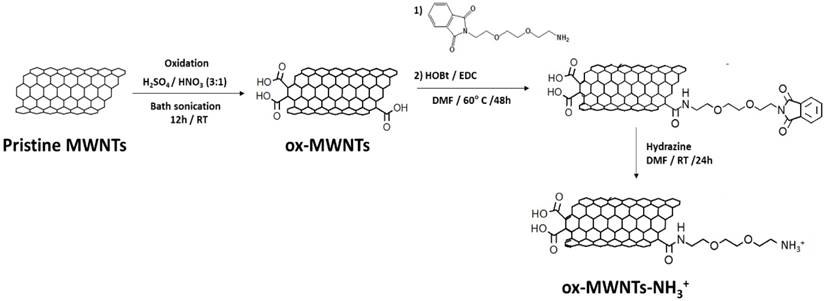
(i) Oxidation: pristine MWNTs (100 mg) were suspended in 25 mL HNO3:H2SO4 (1:3), which was bath sonicated for 12 h (water was regularly changed to prevent overheating). Subsequently, the mixture was added to 500 mL of distilled water (dH2O) and vacuum filtered through a 0.2 μm hydrophilic membrane. The black solid (oxidised MWNTs, ox-MWNTs) was collected from the filter and purified by serial washing/filtration with 300 mL dH2O (1x), 300 mL 5% NaOH (1x) and 200 mL methanol (1x).
(ii) Amidation: ox-MWNTs (~34 mg, 0.076 mmol) were suspended in 40 mL dimethylformamide (DMF) under bath sonication for 10 min and subsequently mixed with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl) (~29.1 mg, 0.152 mmol), N- hydroxybenzotriazole (HoBt) (~20.5 mg, 0.152 mmol) and phthalimide-protected amine-terminated spacer (~42.3 mg, 0.152 mmol; synthesis described in Supplementary Material). The reaction was carried out under stirring at 60°C for 24 h, after which the ox-MWNTs were recovered by vacuum filtration through a 0.2 μm hydrophobic membrane. The collected black solid (amidated ox-MWNTs) was further purified by serial washing/filtration with 100 mL DMF (1x) and 100 mL methanol.
(iii) Deprotection: amidated ox-MWNTs (32 mg) were suspended in 20 mL DMF under bath sonication for 10 min, 6 mL hydrazine monohydrate were then added to the reaction mixture and the suspension was continuously stirred for 24 h at room temperature (RT). The deprotected ox-MWNTs (ox-MWNTs-NH3+) were recovered by vacuum filtration through a 0.2μm hydrophobic filter and further purified via serial washing/filtration with 100 mL DMF (1x) and 100 mL methanol (final yield ~30 mg).
Kaiser test
Ninhydrin assay reagents (phenol/ethanol 42.55 M, KCN/pyridine 0.02 M and ninhydrin/ethanol 0.28 M) were prepared in the fume hood. Five hundred microliters of each reagent were added to 1 mg of ox-MWNTs or ox-MWNTs-NH3+, followed by bath sonication for 1 min and incubation at 100ºC for 7 min. Subsequently, 4.5 mL of ethanol:water (60:40) were added to the suspension, which was further centrifuged for 10 min at 1811 x g (~2000 rpm; Eppendorf 5810R, Fisher Scientific UK). The UV-vis absorbance of the supernatant was measured at 575 nm against a standard curve of 2,2′-(Ethylenedioxy)bis(ethylamine) (Fig. S1) in a Lambda 35 spectrophotometer (Perkin Elmer, USA).
Thermogravimetric (TGA) analysis
Samples of pristine MWNTs, ox-MWNTs and ox-MWNTs-NH3+ (~1 mg) were loaded in platinum pans and the analysis was carried out on a TGA Q500 (TA instruments, USA). Reaction was performed under constant nitrogen gas flow (90 mL/min) and consisted of temperature increase (from RT) and stabilisation at 100◦C for 20 min, followed by a controlled temperature increase (10◦C/min) up to 1000 ◦C. Data analysis was performed using the Advantage software (TA instruments, USA).
Conjugation of L2 and L3 or Gd(L2) and Gd(L3) to f-MWNTs
Gd(L2) and Gd(L3) were conjugated to ox-MWNTs or ox-MWNTs-NH3+ at a molar ratio of 1:10 (complex:f-MWNTs), as shown in Scheme 3. Since it is difficult to calculate the number of benzene rings per f-MWNT, as well as the number of f-MWNTs in each sample, a ratio of 1 molecule of Gd(L2/3) to 10 benzene units was adopted. ox-MWNTs or ox-MWNTs-NH3+ were dispersed in 4 mL dH2O under bath sonication for 5 min and the ligands were added separately to each dispersion, which was briefly sonicated. The different dispersions (ox-MWNTs + Gd(L2), ox-MWNTs + Gd(L3), ox-MWNTs-NH3+ + Gd(L2) and ox-MWNTs-NH3+ + Gd(L3) were continuously stirred (300 rpm) for 48 h at RT. For recovery of the ligand:f-MWNT conjugates, the dispersions were vacuum filtered through 0.2 µm pore hydrophilic membranes, the black solid pellets (conjugates) were collected and purified via serial washing/filtration with 20 mL dH2O (3x), 20 mL methanol:dH2O (1:1) (2x) and 20 mL methanol (2x) (Scheme 3). Samples of the filtrate (supernatant containing unbound ligand) and washing fractions were collected for quantification via inductively coupled plasma mass spectrometry (ICP-MS) and fluorimetry. Samples of conjugates (Gd(L2):ox-MWNTs, Gd(L3):ox-MWNTs, Gd(L2):ox-MWNTs-NH3+ and Gd(L3):ox-MWNTs-NH3+) were collected for quantification of bound ligand via ICP-MS. The same protocol was followed for conjugation of Gd3+-free L2/3 to ox-MWNTs or ox-MWNTs-NH3+.
Conjugation of Gd(L2) or Gd(L3) to f-MWNTs. Gd(L2) or Gd(L3) were conjugated to ox-MWNTs or ox-MWNTs-NH3+ by incubation under stirring for 48 h at RT. To remove the unbound complex, the conjugates (Gd(L2):ox-MWNTs, Gd(L3):ox-MWNTs, Gd(L2):ox-MWNTs-NH3+ and Gd(L3):ox-MWNTs-NH3+) were vacuum filtered through a hydrophilic membrane. The pellet recovered on the filter contains the bound complex, whereas the supernatant (filtrate) contains unbound complex.
Quantification of ligand loading by ICP-MS
ICP-MS quantification of Gd3+ conjugated to f-MWNTs was performed as described previously for Fe3+ conjugated to CNTs [30]. Suitable dilutions of Gd3+ standards (Ricca Chemical, TX, USA) were prepared in 10% nitric acid to obtain a Gd3+ standard curve in the range of 10-3-103 parts per million (ppm) (1 ppm ~103 µg/L ~6.36 µmol/L; MW=157.25 g/mol). To remove any traces of metal, 15 mL non-sterile tubes were pre-treated with 10 mL HCl (0.1 M) at 50°C for 30 min and dried before usage. Samples of conjugates (Gd(L2):ox-MWNTs, Gd(L3):ox-MWNTs, Gd(L2):ox-MWNTs-NH3+, Gd(L3):ox-MWNTs-NH3+), filtrate and washes were digested by incubation with 2 mL HNO3 (67%) for 24 h at 80°C. The resulting solutions were diluted with dH2O to reach a concentration within the calibration range (average final volume of 10 mL) and subsequently centrifuged at 4000 rpm for 30 min (Eppendorf 5810R, Fisher Scientific UK) to ensure the removal of f-MWNTs from the solution. The Gd3+ content in the samples was measured against a standard curve of Gd3+ (Fig. S2) using the NexION 350D ICP mass spectrometer (Perkin Elmer, USA) with Cetac ASX 520 autosampler and Syngistix software. Sample flow was kept at 0.2 mL/min, argon flow (main, aux, nebuliser) at 18, 1.2 and 1.0 L/min, and RF coil power was 1600 W. An average of 5 different replicates was performed per sample measurement (with 50 sweeps/replicate).
Quantification of complex loading by fluorimetry
The excitation and emission spectra of Gd(L2) and Gd(L3) were determined using the LS50B Luminescence Spectrophotometer (Perkin Elmer, USA) (Fig. S3A, B). For sample quantification, 100 µL were loaded onto a black 96-well Costar plate (Corning, USA) and the fluorescence intensity was measured against standard curves of Gd(L2) or Gd(L3) (Fig. S3C, D) with the FLUOstar Omega microplate reader (BMG Labtech, Aylesbury, UK), and a machine gain of 500.
Radiolabelling of L2/3 and L2/3:f-MWNTs conjugates with 111In
Radiolabelling L2 with 111In has been reported previously [11]. In this work, radiolabelling was performed on L2/3:ox-MWNTs and L2/3:ox-MWNTs-NH3+ conjugates with the labelling approach modified accordingly. Dispersions of f-MWNTs (1 mg/ml, in H2O) were mixed with 111InCl3 buffered in ammonium acetate (pH 5.5). Solutions were incubated for 30 min at room temperature and were vortexed every 5 min for thorough mixing. Solutions of L2/3 (1 mg/ml in H2O) were also subjected to the same labelling procedures. The labelling efficiency was assessed by iTLC (mobile phase, 3.5% NH4+: methanol (1:1), pH 11). The auto-radioactivity of each TLC strip was developed and counted quantitatively using a Cyclone phosphor detector (Packard Biosciences). EDTA solution (0.1 M, 1/20, v/v) was then added to the f-MWNTs mixture to chelate any excess unreacted 111InCl3. Labelled 111In(L2/3):f-MWNTs dispersions were pelleted by centrifugation at 13,000 rpm for 30 min while unreacted 111In remained in the supernatant. The pelleted 111In(L2/3):f-MWNTs were resuspended in 1X PBS at 1 mg/ml for injection. iTLC (mobile phase: 0.1 M ammonium acetate buffer containing 50 mM EDTA) was carried out after addition of EDTA and after centrifugation. For labelled 111In(L2/3), solutions were buffered with 10X PBS to become isotonic for injection without the addition of EDTA.
The stability of radiolabelling was examined by incubating 111In(L2/3):f-MWNTs dispersions and 111In(L2/3) solutions with an equal volume of PBS or FBS at 37 °C for 24 h. The solutions were subjected to iTLC analyses as described above.
Different amounts of 111InCl3 were used for radiolabelling for different studies according to the labelling efficiency and radioactivity required. The radioactivity in each injection dose was ~10 MBq and ~1 MBq for SPECT/CT imaging and γ-scintigraphy studies respectively.
Animals
In vivo experiments were conducted under the authority of project and personal licences granted by the UK Home Office and the UKCCCR Guidelines (1998). Female C57BL/6 mice aged 6-8 weeks were purchased from Charles River (UK) for all the experiments. Details about animal husbandry and welfare are provided in Supplementary Material.
SPECT/CT
The biodistribution of intravenously (i.v.)-injected 111In-labelled L2/3, L2/3:ox-MWNTs, and L2/3:ox-MWNTs-NH3+ was firstly examined by 3D whole body SPECT/CT imaging. Mice were injected via tail vein with 100 μg of labelled 111In(L2/3):f-MWNTs (1 mg/ml in PBS) or In(L2/3) alone (100 μg) under isoflurane anaesthesia. SPECT/CT scans were carried out on the same animal at multiple time points: immediately after injection, at 4 h and 24 h. Mice were placed in the prone position and SPECT scans were acquired over 24 projections (60 s per projection), using a 4-head scanner with 1.4 mm pinhole collimators, for a total acquisition time of 30-40 min (Nano-SPECT/CT, USA) [27]. CT scans (45 kVP X-ray) were performed after each SPECT. All images were reconstructed by MEDISO software (Medical Imaging Systems), and SPECT and CT images were merged using the VivoQuantTM software (inviCRO, USA).
γ-scintigraphy
Quantitative organ uptake of i.v. injected 111In-labelled L2/3 and L2/3:ox-MWNTs-NH3+ was assessed by γ-scintigraphy. Mice were injected via a tail vein with 111In(L2/3):ox-MWNTs-NH3+ (50 μg per mouse) or 111In(L2/3) (50 μg per mouse) under isoflurane anaesthesia. At 2, 5, 10 and 30 min after injection, blood samples were collected from the lateral tail vein and the mice were perfused with 50 mL of heparinised saline solution (50 U/mL) through the left ventricle of the heart to wash out the 111In-labelled compounds remaining in circulation or loosely attached to tissues. Major organs (including brain) were excised, weighed and the radioactivity was measured by gamma counting (LKB Wallac 1282 Compugamma, PerkinElmer). Results (mean ± SD, n=3) are expressed as % of injected dose per gram of organ (% ID/ g of organ), unless otherwise stated.
Capillary depletion
Brain tissues were subjected to capillary depletion, i.e. separation of brain parenchyma from blood vessels (capillaries), as described previously [31]. Briefly, each brain was homogenised in a glass homogeniser containing 1 mL of ice-cold depletion buffer (10 mM HEPES in HBSS, pH 7.4) via 15 stokes of the pestle, followed by addition of 1.6 mL of depletion buffer containing 26% dextran (148 kDa) and repeated pestle homogenisation (3 strokes). The homogenates were centrifuged at 3220 x g (Eppendorf 5810R, Fisher Scientific UK) for 15 min to separate parenchyma (supernatant) and capillaries (pellet), and the radioactivity was measured using gamma counting (LKB Wallac 1282 Compugamma, PerkinElmer). Results (mean ± SD, n=3) are expressed as % of injected dose per brain fraction (% ID/brain fraction) followed by the analysis of brain parenchyma to blood ratio.
Statistical analysis
Data are presented as mean ± standard deviation (mean ± SD) of at least two different experiments, unless otherwise stated. Statistical analysis was performed on GraphPad Prism (GraphPad Software, CA, USA) using one-way ANOVA with Tukey's multi-comparison test, unless otherwise stated.
Results and discussion
Synthesis and characterisation of Gd(L2), Gd(L3) and f-MWNTs
The synthesis of the ligand L2 was previously reported and the synthetic pathway of L3 is outlined in Scheme 1. 4-(6-methoxybenzo[d]thiazol-2-yl)aniline 1 was prepared according to the procedures described in the literature [12]. Then, in a reductive amination reaction, the N-dimethylated derivative 2 was obtained in very high yield, and the ether function was quantitatively cleaved with boron tribromide. The linker was then introduced by nucleophilic substitution between product 3 and N-(6-Bromohexyl)phthalimide and the removal of the phtalimide protecting group was performed with hydrazine hydrate, giving the corresponding amine 5, which was acylated with chloroacetyl chloride to afford the amide derivative 6. Protected compound 7 was obtained by N-alkylation of tri-tert-butyl 2,2',2''-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (DO3A-tBu) with 6 under classical conditions in presence of a base, in acetonitrile. The acid-sensitive tert-butyl protecting groups were then removed with trifluoroacetic acid in dichloromethane to give the final ligand L3. The gadolinium complexes Gd(L2) and Gd(L3) were formed by adding the gadolinium salt at a controlled pH of 5,5.
Since the conjugation (via adsorption) of ligands on MWNTs is dependent on the hydrophobicity of the probes, an increase in the log P values of the ligands could be beneficial. The ligand structures diverge on the spacer used for conjugation of the PiB derivative to Do3A, with amide or ether linkages being used in L2 and L3, respectively. The lipophilicity of L3/ Gd(L3) was also modulated by introducing two methyl groups on the amine function. Consequentially, the log P value of Gd(L3) shows a gain in lipophilicity (Log P = 0.63) compared to Gd(L2) (Log P = 0.03).
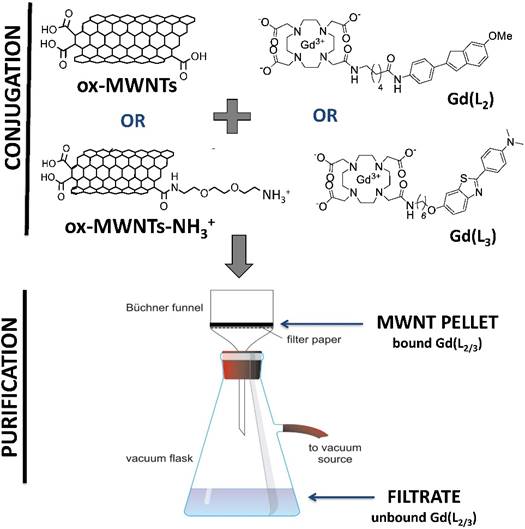
Due to high hydrophobicity, pristine MWNTs are not easily dispersed in water. Moreover, the reduced diameter (~9.5 nm) of the MWNTs used in this study makes them prone to aggregation in aqueous solution [32]. For this reason, pristine MWNTs were initially oxidised by incubation (under sonication) with a mixture of sulphuric and nitric acid, to promote chemical exfoliation of the nanotube surface, addition of carboxylic (COOH) groups and, consequently, increasing water dispersibility and decreasing possible cell toxicity (Scheme 2). Functionalisation via oxidation has also been shown to enhance renal clearance and decrease reticuloendothelial system accumulation [33]. The presence of carboxylate groups on the side-wall defects and tips of MWNTs also allows for introduction of additional functional groups [34]. Indeed, oxidised MWNTs (ox-MWNTs) were further functionalised via amidation reaction between COOH groups and a phthalimide-protected amine-terminated spacer (Scheme 2). Deprotection of the spacer was achieved by alkaline treatment, yielding ox-MWNTs with positively charged primary amine groups (ox-MWNTs-NH3+). The introduction of functional groups to the surface of pristine MWNTs was confirmed by TGA and Kaiser test. TGA allows accurate determination of MWNT functionalisation by assessing sample weight loss upon exposure to gradually increasing temperature (under inert atmosphere). The weight loss is directly related to the introduction of functional groups onto the side-walls of f-MWNTs. As shown in Fig. 2A, pristine MWNTs exhibit high thermal stability (i.e. residual weight loss) up to 700 °C, after which the benzene backbone starts to decompose. In comparison, substantial weight loss was detected for ox-MWNTs at 600 °C (~13.5%), indicating the thermal degradation of carboxylic groups. Additional weight loss was detected for the amidated ox-MWNTs at 600 °C (~9.2%, total mass loss = 22.7%), which confirms the degradation of amine groups on the surface of ox-MWNTs-NH3+.
Characterisation of f-MWNT functionalization using TGA and Kaiser test. (A) Thermal decomposition of pristine, oxidised (ox-MWNTs) or oxidised and aminated carbon nanotubes (ox-MWNTs-NH3+) under increasing temperature. Functional groups present on f-MWNTs, such as COOH and NH3+, are degraded at temperatures below 600°C (which is reflected by the increasing weight loss), whereas no significant degradation (i.e. residual weight loss) is seen with pristine MWNTs. (B) Representative image of the colorimetric detection of free primary amine groups on the surface of MWNTs (via Kaiser test). No color development is seen with ox-MWNTs (1 mg) (left), whereas development of pink coloration is seen with ox-MWNTs-NH3+ (1 mg) (right).
Surface functionalisation with amine groups was further confirmed using Kaiser test, a semi-quantitative colorimetric test based on the reaction of the ninhydrin reagent with free primary amine groups [35]. Fig. 2B shows a representative assay image, with the development of pink colour (i.e. presence of amine groups) for ox-MWNTs-NH3+, while no colour development is seen for ox-MWNTs (do not contain primary amine groups). An average of 203 µmol of amine groups per gram of ox-MWNTs-NH3+ was obtained using the Kaiser test, a value considerably smaller than that determined by TGA (687 µmol NH2/g of ox-MWNTs-NH3+) (Table 1). This difference is most probably caused by residual aggregation of ox-MWNTs-NH3+, which limits the number of amine groups available to react with the ninhydrin reagent [25, 35]. The amine content of ox-MWNTs-NH3+ used this study is nevertheless similar to that reported in previous studies from our group, with values ranging from 100 to 530 µmol of amine groups per gram of MWNTs generated by amidation reaction [25, 26, 29, 34].
Quantitative characterisation of MWNT functionalisation by TGA and Kaiser test.
| f-MWNTs | μmol of functional group per gram of MWNTs | ||
|---|---|---|---|
| TGA (a) | Kaiser test (b) | ||
| ox-MWNTs | 2446 ± 770 | - | COOH |
| ox-MWNT-NH3+ | 682 ± 10 | 203 ± 21 | NH2 |
(a) Measured using TGA.
(b) Measured by Kaiser test.
Taking into consideration the extent of MWNT oxidation determined by TGA (2446 ± 770 µmol COOH/ g of ox-MWNTs), the data indicates that ~1/3 of the carboxylate groups were functionalised with the amine-terminated linker. The size of the f-MWNTs has been studied previously, with the mean diameter of ox-MWNTs measured between 6 and 12 nm and the mean length between 370 and 570 nm, by transmission electron microscopy and subsequent image analysis with Image J [26, 33]. The amidation reaction and conjugation of ligands are not expected to affect nanotube length, due to absence of oxisiding acid in this step. Similar to what has been observed in our previous studies [25, 26, 33, 34], good water dispersibility was obtained for both ox-MWNTs and ox-MWNTs-NH3+ (data not shown).
Conjugation of Gd(L2) or Gd(L3) to f-MWNTs
Since Gd(L2)/Gd(L3) contain aromatic rings in their structure, the conjugation to ox-MWNTs or ox-MWNTs-NH3+ - by simple mixing under stirring - relied on hydrophobic interactions between the ligands and MWNTs. Conjugation was assessed by two complementary techniques, ICP-MS and fluorimetry, upon separation of MWNT-bound and unbound ligand fractions by vacuum filtration (Scheme 3). ICP-MS was used to quantify Gd3+ in MWNT-bound and unbound fractions, providing an indirect measurement of Gd(L2/3) conjugated to the MWNTs. Taking advantage of the intrinsic fluorescence of L2/3, fluorimetry was used to quantify the ligands in the unbound fraction, while quantification of the MWNT-bound fraction by this technique is hampered by the fluorescence-quenching properties of the MWNTs.
As shown in Table 2, higher levels of Gd(L2) and Gd(L3) were found conjugated to ox-MWNTs by ICP-MS (389 ± 438 and 222 ± 70 μmol of complex/g of f-MWNTs, respectively) compared to ox-MWNT-NH3+ (60 ± 14 and 105 ± 33 μmol of complex/g of f-MWNTs). Similarly, significantly higher levels of Gd(L2/3) were found conjugated to ox-MWNTs when loading was measured by fluorimetry (p=0.0071, two-way ANOVA). The favourable ligand loading onto ox-MWNTs is probably related to the steric hindrance generated by the amine-terminated spacer, which partly blocks the interaction between aromatic rings of ligands and f-MWNTs. A trend towards more efficient conjugation of Gd(L2) was also detected by both ICP-MS and fluorimetry (389 ± 438 and 542 ± 42 µmol of ligand/g of f-MWNTs, respectively), when compared to Gd(L3) (222 ± 70 and 203 ± 2 µmol of ligand/g of f-MWNTs) (p=0.54 for ICP-MS, p=0.0069 for fluorimetry; two-way ANOVA). This observation could possibly be justified by the difference in hydrophobicity of Gd(L2) and Gd(L3) (log P = 0.03 and 0.63, respectively).
Quantification of ligand loading onto f-MWNTS by ICP-MS and fluorimetry.
| f-MWNTs | μmol of ligand per gram of f-MWNTs (a) | |||
|---|---|---|---|---|
| Gd(L2) | Gd(L3) | |||
| ICP-MS | Fluorimetry (c) | ICP-MS (b) | Fluorimetry | |
| ox-MWNTs | 389 ± 438 | 542 ± 42 | 222 ± 70 | 203 ± 2 |
| ox-MWNT-NH3+ | 60 ± 14 | 577 ± 8 | 105 ± 33 | 51 ± 29 |
a) Values are presented as mean ± SD.
b) Measured by ICP-MS.
c) Obtained by subtracting the unbound fraction (measured by fluorimetry) from the total ligand in reaction.
Radiolabelling of L2/3 and L2/3:f-MWNTs conjugates with 111In
Since the PiB-DO3A derivatives have high affinity for both gadolinium(III) and indium(III), (L2/3):f-MWNTs conjugates prepared without Gd3+ were radiolabelled with 111In3+, aiming at assessing the in vivo biodistribution and brain uptake of the constructs by SPECT/CT imaging and γ-scintigraphy with high sensitivity. Although indirect tracking of the f-CNT has been carried out in this study, it is worth reporting that label-free detection of f-CNTs in vivo has been previously reported by our group. Taking advantage of the optical properties of CNTs, we used Raman and multiphoton luminescence to confirm the direct presence of systemically-administered f-MWNTs in mouse brain [27]. Furthermore, the presence of CNTs was confirmed by TEM in both whole brain and capillary fractions after intravenous injection in mice [27]. The results matched the indirect detection carried out by radiolabelling. We therefore adapted radiolabelling method to confirm the conjugation and track the organ biodistribution of L2 and L3 ligands.
The labelling efficiency was determined by iTLC analyses. Mobile phase consisting of 3.5 % NH4+:methanol (1:1) was used to test labelling efficiency of L2/3. As shown in Fig. 3A, labelled conjugates (111In(L2/3)) were detected at the solvent front (labelling efficiency > 60%). To test the labelling efficiency of (L2/3):f-MWNTs conjugates, a mobile buffer containing 50mM EDTA in 0.1 M acetate buffer was used, with an average labelling efficiency > 85% (spots at application point) being determined (Fig. 3B). Removal of unconjugated 111In (in the form of [111In]EDTA) or [111In]L2/3) from the final sample was achieved by centrifugation.
Labelling efficiency and stability studies of 111In-labelled L2/3 and their f-MWNTs conjugates. Radiolabelling of (A) L2/3 (B) L2/3:ox-MWNTs and L2/3:ox-MWNTs-NH3+ with 111In3+ was determined by iTLC analyses using 3.5 % NH4+: methanol (1:1, pH 11) (for L2/3) and 0.1 M ammonium acetate buffer containing 50 mM EDTA (pH 5.5) (for the conjugates) as the mobile phase. Dispersions of 111In(L2/3):f-MWNTs were treated with 0.1 M EDTA (1/20 v/v) prior TLC run to chelate unreacted 111In. (C) After centrifugation and re-dispersion in PBS, dispersions were incubated with or without FBS (1:1, v/v) at 37 °C for 24 h. The labelling stability was examined by iTLC (mobile phase: 0.1 M ammonium acetate buffer containing 50 mM EDTA).
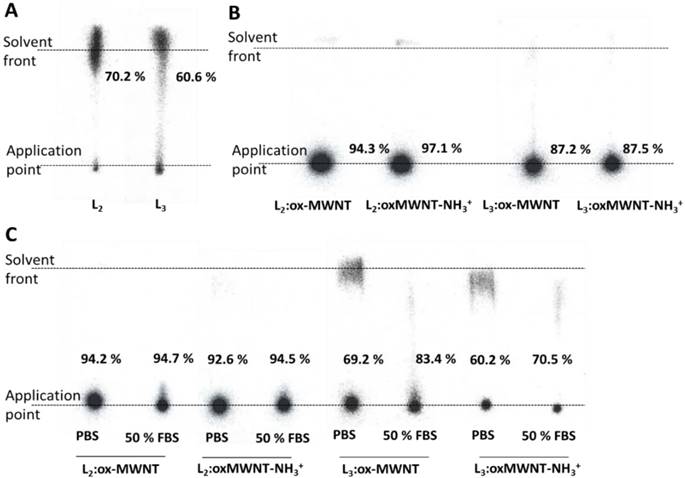
The labelling stability was assessed upon incubation in PBS or 50% FBS at 37 °C for 24 h. The iTLC results revealed that greater radiochemical stability was obtained for 111In(L2):f-MWNTs than 111In(L3):f-MWNTs conjugate. The presence of serum had no influence on the stability (Fig. 3C).
Whole body SPECT/CT imaging of 111In-labelled conjugates following i.v. injection
Whole body SPECT/CT imaging of 111In-labelled free ligands (L2/3) and (L2/3):f-MWNTs conjugates was performed to monitor the spatial distribution of the compounds upon i.v. administration in healthy mice. As shown in Fig. 4, marked differences in biodistribution were observed between 111In(L2/3) and the (L2/3):f-MWNTs conjugates.
Whole body SPECT/CT imaging of 111In-labelled L2/3 and L2/3:f-MWNTs conjugates following i.v. injection. C57BL/6 mice were injected via tail vein with ~10 MBq of non-conjugated complexes 111In(L2/3) (100 µg) and their f-MWNTs conjugates 111In(L2/3):ox-MWNTs and 111In(L2/3):ox-MWNTs-NH3+ (200 µg). SPECT/CT imaging was carried out immediately after injection and at 4 and 24 h post injection.
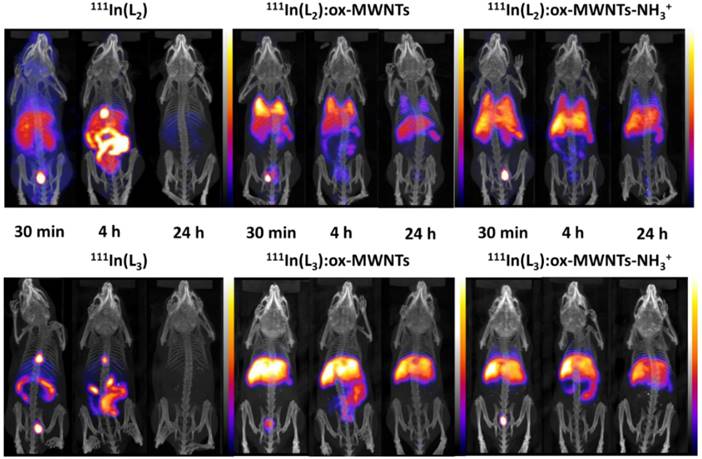
The SPECT/CT images at 30 min and 4 h post injection suggested that 111In(L2/3) underwent fast renal excretion as indicated by the strong signals detected in bladder and/or kidney. Biliary excretion was also observed evidenced by the intense signals in gallbladder and gut lumen within the first 4 h after injection.
Unlike the free metal complexes, typical distribution patterns of 111In:f-MWNTs [26, 34] were obtained for the studied 111In(L2/3):f-MWNTs, indicating the successful conjugation of (L2/3) with f-MWNTs successful in chelating 111In. The biodistribution profiles were essentially established at the early time point for all the conjugates. 111In(L2):ox-MWNTs showed accumulation in the order of lung>liver=spleen while 111In(L2):ox-MWNTs-NH3+ displayed comparable levels of uptake in these three organs. Reduction in lung uptake was observed for both 111In(L2):f-MWNTs over time. In contrast to 111In(L2):f-MWNTs, most of 111In(L3):f-MWNTs was detected in liver and, at a smaller extent, spleen (up to 24 h). Some signals were detected in the bladder/kidney (30 min) and in the gut lumen (4 h) suggesting dissociation of 111In(L2/3) from the conjugates.
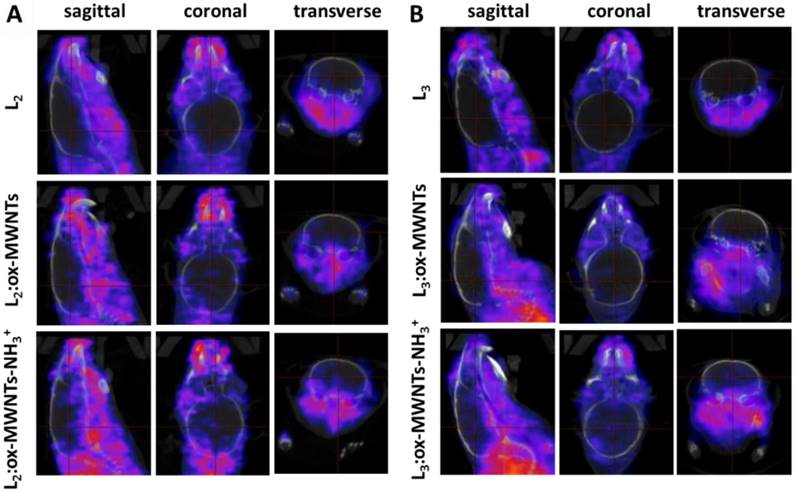
SPECT/CT images focusing on the brain at the early time point (30 min) are shown in Fig. 5. Signals of 111In(L2/3) were excluded from the brain cavity while some signals of 111In(L2/3):f-MWNTs can be seen inside and brain. 111In(L2/3):ox-MWNTs-NH3+ showed higher brain accumulation than 111In(L2/3):ox-MWNTs with the highest observed for 111In(L2):ox-MWNTs-NH3+.
SPECT/CT images of 111In-labelled L2/3 and L2/3 :f-MWNTs conjugates in mouse brain following i.v. injection. C57BL/6 mice were i.v. injected with (A) 111In(L2), 111In(L2):ox-MWNTs, and 111In(L2):ox-MWNTs-NH3+ and (B) 111In(L3), 111In(L3):ox-MWNTs, and 111In(L3):ox-MWNTs-NH3+, via a tail vein. SPECT/CT imaging was performed within 30 min post injection.
Quantitative assessments of organ uptake of 111In(L2/3) and 111In(L2/3):ox-MWNTs-NH3+ following i.v. injection by γ-scintigraphy
Quantitative organ biodistribution and brain uptake was performed by γ-scintigraphy for amine modified derivatives as they showed higher brain uptake than the carboxylated derivatives; 111In(L2/3) were used for comparison.
As revealed in Fig. 6A, significantly higher levels of 111In(L2/3) were found in the blood 2 min after injection (~44.0-49.1 % ID) compared to MWNTs conjugates (~12.1-15.3 % ID) (p<0.001). The prolonged blood circulation time observed for 111In(L2/3) was possibly due to binding to serum proteins. The short blood circulation times detected for f-MWNTs conjugates agreed with our previous studies with values ranging from 6 to 14 % of ID at 2-5 min post-injection [25, 27].
Blood and organ biodistribution profiles of 111In-labelled L2 and L3, in its free form or conjugated to ox-MWNTs-NH3+, following systemic administration in healthy C57BL/6 mice. Mice were injected (via tail vein) with 111In-labelled L2/3 or 111In(L2/3):ox-MWNTs-NH3+ (50 μg, 0.5 MBq). Blood was sampled at 2, 5, 10 and 30 min post-injection, followed by whole-body perfusion with heparinised saline. All major organs were immediately harvested and the radioactivity of (A) blood and (B) organs was measured by gamma counting. The Results are presented as (A) % injected dose per blood and (B) % injected dose per gram of tissue (inset: magnification of brain distribution). Statistical analysis of the results (mean ± SD, n = 3;) was performed using One-way ANOVA with Tukey's posthoc test. (A) *** p<0.001 compared to animals injected with 111In(L2/3):ox-MWNTs-NH3+ and ## p<0.01 compared to animals injected with 111In(L2/3):ox-MWNTs-NH3+; (B) *** p<0.001 compared to animals injected with 111In(L2/3) or 111In(L3):ox-MWNTs-NH3+; ## p<0.01 compared to animals injected with 111In(L2/3) or 111In(L3):ox-MWNTs-NH3+; + p<0.05, +++ p<0.001 compared to animals injected with 111In(L2):ox-MWNTs-NH3+ or L2/3, respectively; ^^ p<0.01, ^^^ p<0.001 compared to animals injected with 111In(L2):ox-MWNTs-NH3+ or 111In(L3):ox-MWNTs-NH3+.
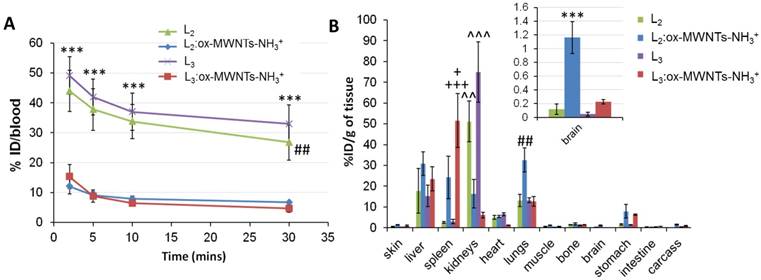
The analysis of the biodistribution profile of conjugates/complexes (Fig. 6B) revealed that 111In(L2/3):ox-MWNTs-NH3+ accumulated primarily in the liver, spleen and lungs 30 min after injection, with differences between the two conjugates: while similar levels were detected in the liver (~23.5-30.9 %ID/g of tissue), preferential lung accumulation was found for 111In(L2):ox-MWNTs-NH3+ (32.6 ± 5.8%ID/g of tissue) than 111In(L3):ox-MWNTs-NH3+ (12.7 ± 2.3 %ID/g of tissue) (p<0.01). On the contrary, higher spleen uptake was found for 111In(L3):ox-MWNTs-NH3+ (51.6 ± 12.9) than 111In(L2):ox-MWNTs-NH3+ (24.2 ± 10.3 %ID/g) (p<0.05). Uptake of f-MWNT in RES organs was previously reported in our studies [25-27, 34]. Agreeing with SPECT/CT data, 111In(L2/3) showed significantly higher levels in the kidneys (~51.1- 74.9 %) compared to the conjugates (~6.1-16.3).
The preferential accumulation of L2 and L3 conjugates in the lung and spleen, respectively, could be due to difference in structure of possibly the final net charge of the conjugates. It has been shown in pre- clinical studies involving lipid-mediated delivery of siRNAs to dendritic cells that adjusting nanoparticle net charge to negative values can benefit spleen uptake, whereas positively-charged nanoparticles favour lung retention [36]. Although the physico-chemical, structural and biological properties of f-MWNTs and lipid nanoparticles are significantly different, this observation could help explain the differential uptake of 111In(L2/3):ox-MWNTs-NH3+ by lungs and spleen. Since L2 and L3 are probably neutrally charged at physiological pH, the determination of the net surface charge of 111In(L2/3):ox-MWNTs-NH3+ complexes in the blood could is needed to help understand this observation.
Importantly, previous histological studies from our group using organ samples from mice injected intravenously with ox-MWNTs-NH3+ showed no signs of tissue necrosis, fibrosis, or inflammation at different time points after administration (1h, 7 days, 30 days) [33]. Moreover, in vivo degradation of CNTs has been demonstrated in different studies. Using TEM and Raman spectroscopy, our group found evidence of MWNT-NH3 + degradation in mouse brain 14 days after stereotactic injection, with partial degradation following internalization and intracellular localization within microglia starting as early as 2 days after injection [28]. Studies by Kagan and colleagues using TEM, Raman spectroscopy and photoacoustic imaging have also shown peroxynitrite-induced macrophage-mediated degradation of SWNTs in lung tissue following pharyngeal administration in mice [37]. Nevertheless, further studies need to provide qualitative and quantitative data regarding the formation of by-products induced by CNT degradation, aiming at its clinical application.
Fig. 6B revealed higher brain accumulation of 111In(L2):ox-MWNTs-NH3+ (1.16 ± 0.23 %ID/g) than 111In(L3):ox-MWNTs-NH3+ (0.22 ± 0.03 %ID/g (p<0.001) or 111In(L2/3) (~0.07-0.12 %ID/g) (p<0.001) at 30 min post-injection. This value is superior to those reported for 111In-labelled L1 (0.36 and 0.5 %ID/g tissue in cortex and cerebellum at 30 min) [11], PiB [38] or Aβ-targeting ApoE-conjugated phosphatidic acid-encapsulating liposomes (a maximum of 0.3% ID/g of brain achieved at 24h after injection) [39].
Brain distribution profile of 111In(L2/3) and 111In(L2/3):ox-MWNTs-NH3+ following i.v. injection by γ-scintigraphy
To determine the amount of injected conjugates/ligands that reach the different brain tissues, endothelial cells (capillaries) and parenchyma were separated using capillary depletion (Fig. 7A). Higher levels of 111In(L2):ox-MWNTs-NH3+ were detected in both parenchyma and capillary fractions (~0.15 and 0.26 %ID/fraction, respectively) than 111In(L3):ox-MWNTs-NH3+ (~0.04 and 0.09 %ID/fraction, p<0.001 and 0.01). While a larger proportion of 111In(L2):ox-MWNTs-NH3+ was found in capillaries (~0.26 %ID), the difference to the parenchyma fraction was not statistically significant (~0.15 %ID; p>0.05 using unpaired t-test with Welch's correction). Values detected for 111In(L2/3) were all below 0.05 %ID/fraction.
Brain distribution profile of 111In-labelled L2 and L3, in its free form or conjugated to ox-MWNTs-NH3+, following systemic administration in healthy C57BL/6 mice. Mice were injected (via tail vein) with 111In-labelled L2/3 or 111In(L2/3):ox-MWNTs-NH3+ (50 μg, 0.5 MBq). After measuring the radioactivity of the whole organs, brain tissues were processed to separate brain parenchyma from capillaries (via capillary depletion) and the radioactivity of each fraction was measured by gamma counting (A), with the data presented as % ID per total brain parenchyma or capillaries. (B) For brain parenchyma to blood ratio calculation, the weight of parenchyma was considered as the weight of the whole brain. Inset: magnification of brain parenchyma/blood ratio for L2 and L3. Statistical analysis of the results (mean ± SD, n = 3;) was performed using unpaired t-test with Welch's correction. (A) *** p<0.001 compared to parenchyma of animals injected with 111In(L2/3) or 111In(L3):ox-MWNTs-NH3+ and ## p<0.01 compared to capillaries of animals injected with 111In(L2/3) or 111In(L3):ox-MWNTs-NH3+ (B) ** p<0.01 compared to animals injected with 111In(L3):ox-MWNTs-NH3+ and ## p<0.01 compared to animals injected with 111In(L2/3).
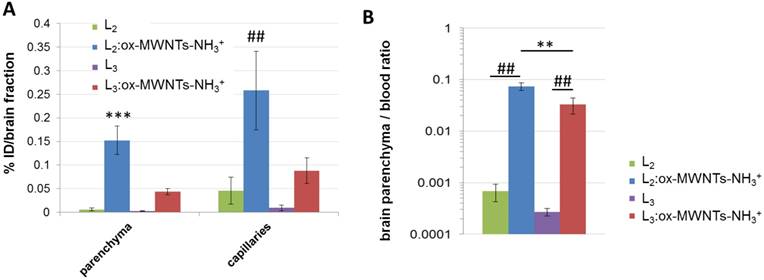
A better measure for the extent of brain penetration - i.e. the translocation from blood to brain tissue across the BBB - is given by the brain parenchyma to blood ratio (P/B ratio) [40]. In this regard, the highest P/B ratio was obtained for 111In(L2):ox-MWNTs-NH3+ (~0.074) compared to 111In(L3):ox-MWNTs-NH3+ (~0.04, p<0.01) or to the residual values obtained for 111In(L2/3) (Fig. 7B).
Overall, these results indicate that conjugation of L2 to ox-MWNTs-NH3+ significantly improved its brain accumulation while a small and non-significant improvement was obtained for L3. The whole-brain uptake values (~1.16 % ID/g of tissue) and P/B ratio (~0.074) obtained in this study were lower than those obtained in previous studies from our group (1.5 to 3% ID/g of brain; 0.2 to 0.7 P/B ratio) [26, 39] and those of known CNS drugs (including morphine) [41] (P/B ratios > 0.5). They are however higher than those reported for other types of brain-targeting nanoparticles (0.3 to 0.5% ID/g of brain) [31, 42-44] and several clinical CNS drugs (~0.2% of administered dosage) [45-47]. Future conjugation to brain-targeting peptides (e.g. angiopep-2, a ligand of low-density lipoprotein receptor-related protein 1 (LPR-1)) may improve brain targeting even further [26].
Conclusion
This study provides evidence of the potential of functionalised MWNTs to be used as carriers for brain delivery of drugs/compounds that do not significantly cross the BBB, with possible clinical application in the diagnostic and treatment of neurological disorders. Future studies will aim at improving brain uptake of Gd(L2):MWNT conjugates via receptor-mediated transcytosis (by conjugation to angiopep1) and further testing the imaging and therapeutic ability in a relevant AD mouse model.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors would like to thank Andy Cakebread (King's College London) for the assistance with the ICP-MS measurements and Zoltan Garda and Alexandre C. Oliveira (CBM Orléans) for the logP measurements. This work was supported by funds from the Wellcome Trust [WT103913], Biotechnology and Biological Sciences Research Council [BB/J008656/1], Worldwide Cancer Research [12-1054], Brain Tumour Charity [GN-000398], the Ligue contre le Cancer Grand-Ouest [comittees 17 and 45] and the Agence Nationale de la Recherche, France [ANR-16-CE18-0022-01] and COST Action TD1004. P. M. Costa is a Sir Henry Wellcome Post-doctoral fellow.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2011;1:a006189
2. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nature Immunology. 2015;16:229-36
3. Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer's disease. Neuropeptides. 2015;52:1-18
4. Dubois B, Picard G, Sarazin M. Early detection of Alzheimer's disease: new diagnostic criteria. Dialogues in Clinical Neuroscience. 2009;11:135-9
5. Forlenza OV, Radanovic M, Talib LL, Aprahamian I, Diniz BS, Zetterberg H. et al. Cerebrospinal fluid biomarkers in Alzheimer's disease: diagnostic accuracy and prediction of dementia. Alzheimer's & Dementia. 2015;1:455-63
6. Ono M, Wilson A, Nobrega J, Westaway D, Verhoeff P, Zhuang ZP. et al. 11C-labeled stilbene derivatives as Abeta-aggregate-specific PET imaging agents for Alzheimer's disease. Nuclear Medicine and Biology. 2003;30:565-71
7. Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT. et al. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorganic & Medicinal Chemistry Letters. 2002;12:295-8
8. The chemistry of contrast agents in medical magnetic resonance imaging. 2nd ed: John Wiley & Sons, Ltd; 2013.
9. Yang L, Chen Q, Liu Y, Zhang J, Sun D, Zhou Y. et al. Se/Ru nanoparticles as inhibitors of metal-induced AB aggregation in Alzheimer's disease. Journal of Materials Chemistry B. 2014;2:1977-1987
10. Yin T, Yang L, Liu Y, Zhou X, Sun J, Lin J. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer's disease. Acta Biomaterialia. 2015;25:172-183
11. Martins AF, Morfin JF, Kubickova A, Kubicek V, Buron F, Suzenet F. et al. PiB-Conjugated, Metal-Based Imaging Probes: Multimodal Approaches for the Visualization of beta-Amyloid Plaques. ACS Medicinal Chemistry Letters. 2013;4:436-40
12. Martins AF, Morfin JF, Geraldes CF, Toth E. Gd(3+) complexes conjugated to Pittsburgh compound B: potential MRI markers of beta-amyloid plaques. Journal of Biological Inorganic Chemistry. 2014;19:281-95
13. Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF. et al. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12462-7
14. Nau R, Sorgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clinical Microbiology Reviews. 2010;23:858-83
15. Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541-53
16. Pardridge WM. Targeting neurotherapeutic agents through the blood-brain barrier. Archives of Neurology. 2002;59:35-40
17. Lay CL, Liu HQ, Tan HR, Liu Y. Delivery of paclitaxel by physically loading onto poly(ethylene glycol) (PEG)-graft-carbon nanotubes for potent cancer therapeutics. Nanotechnology. 2010;21:065101
18. Zhu X, Xie Y, Zhang Y, Huang H, Huang S, Hou L. et al. Thermo-sensitive liposomes loaded with doxorubicin and lysine modified single-walled carbon nanotubes as tumor-targeting drug delivery system. Journal of Biomaterials Applications. 2014;29:769-79
19. Pantarotto D, Singh R, McCarthy D, Erhardt M, Briand JP, Prato M. et al. Functionalized carbon nanotubes for plasmid DNA gene delivery. Angewandte Chemie. 2004;43:5242-6
20. Vardharajula S, Ali SZ, Tiwari PM, Eroglu E, Vig K, Dennis VA. et al. Functionalized carbon nanotubes: biomedical applications. International Journal of Nanomedicine. 2012;7:5361-74
21. Dumortier H, Lacotte S, Pastorin G, Marega R, Wu W, Bonifazi D. et al. Functionalized carbon nanotubes are non-cytotoxic and preserve the functionality of primary immune cells. Nano Letters. 2006;6:1522-8
22. Farvadi F, Tamaddon A, Sobhani Z, Abolmaali SS. Polyionic complex of single-walled carbon nanotubes and PEG-grafted-hyperbranched polyethyleneimine (PEG-PEI-SWNT) for an improved doxorubicin loading and delivery: development and in vitro characterization. Artificial Cells, Nanomedicine, and Biotechnology. 2016:1-9
23. Wolski P, Nieszporek K, Panczyk T. Pegylated and folic acid functionalized carbon nanotubes as pH controlled carriers of doxorubicin. Molecular dynamics analysis of the stability and drug release mechanism. Physical Chemistry Chemical Physics. 2017;19:9300-12
24. Costa PM, Bourgognon M, Wang JT, Al-Jamal KT. Functionalised carbon nanotubes: From intracellular uptake and cell-related toxicity to systemic brain delivery. Journal of Controlled Release. 2016;241:200-19
25. Kafa H, Wang JT, Rubio N, Venner K, Anderson G, Pach E. et al. The interaction of carbon nanotubes with an in vitro blood-brain barrier model and mouse brain in vivo. Biomaterials. 2015;53:437-52
26. Kafa H, Wang JT, Rubio N, Klippstein R, Costa PM, Hassan HA. et al. Translocation of LRP1 targeted carbon nanotubes of different diameters across the blood-brain barrier in vitro and in vivo. Journal of Controlled Release. 2016;225:217-29
27. Wang JT, Rubio N, Kafa H, Venturelli E, Fabbro C, Menard-Moyon C. et al. Kinetics of functionalised carbon nanotube distribution in mouse brain after systemic injection: Spatial to ultra-structural analyses. Journal of Controlled Release. 2016;224:22-32
28. Nunes A, Bussy C, Gherardini L, Meneghetti M, Herrero MA, Bianco A. et al. In vivo degradation of functionalized carbon nanotubes after stereotactic administration in the brain cortex. Nanomedicine. 2012;7:1485-94
29. Hassan HA, Smyth L, Rubio N, Ratnasothy K, Wang JT, Bansal SS. et al. Carbon nanotubes' surface chemistry determines their potency as vaccine nanocarriers in vitro and in vivo. Journal of Controlled Release. 2016;225:205-16
30. Bai J, Wang JT, Rubio N, Protti A, Heidari H, Elgogary R. et al. Triple-Modal Imaging of Magnetically-Targeted Nanocapsules in Solid Tumours In Vivo. Theranostics. 2016;6:342-56
31. van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM. Comparison of five different targeting ligands to enhance accumulation of liposomes into the brain. Journal of Controlled Release. 2011;150:30-6
32. Chang X, Henderson WM, Bouchard DC. Multiwalled carbon nanotube dispersion methods affect their aggregation, deposition, and biomarker response. Environmental Science & Technology. 2015;49:6645-53
33. Al-Jamal KT, Nunes A, Methven L, Ali-Boucetta H, Li S, Toma FM. et al. Degree of chemical functionalization of carbon nanotubes determines tissue biodistribution and excretion profile. Angewandte Chemie International Edition. 2012;51:6389-6393
34. Wang JT, Fabbro C, Venturelli E, Menard-Moyon C, Chaloin O, Da Ros T. et al. The relationship between the diameter of chemically-functionalized multi-walled carbon nanotubes and their organ biodistribution profiles in vivo. Biomaterials. 2014;35:9517-28
35. Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Analytical Biochemistry. 1970;34:595-8
36. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396-401
37. Kagan VE, Kapralov AA, St Croix CM, Watkins SC, Kisin ER, Kotchev GP. et al. Lung macrophages “digest” carbon nanotubes using superoxide/peroxynitrite oxidative pathway. ACS Nano. 2014;8:5610-21
38. Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. Journal of Medicinal Chemistry. 2003;46:2740-54
39. Bana L, Minniti S, Salvati E, Sesana S, Zambelli V, Cagnotto A. et al. Liposomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Abeta aggregation features and cross the blood-brain-barrier: implications for therapy of Alzheimer disease. Nanomedicine. 2014;10:1583-90
40. van Rooy I, Cakir-Tascioglu S, Hennink WE, Storm G, Schiffelers RM, Mastrobattista E. In vivo methods to study uptake of nanoparticles into the brain. Pharmaceutical Research. 2011;28:456-71
41. Hammarlund-Udenaes M, Friden M, Syvanen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharmaceutical Research. 2008;25:1737-50
42. Ke W, Shao K, Huang R, Han L, Liu Y, Li J. et al. Gene delivery targeted to the brain using an angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials. 2009;30:6976-85
43. Schnyder A, Krahenbuhl S, Drewe J, Huwyler J. Targeting of daunomycin using biotinylated immunoliposomes: pharmacokinetics, tissue distribution and in vitro pharmacological effects. Journal of Drug Targeting. 2005;13:325-35
44. Xie Y, Ye L, Zhang X, Cui W, Lou J, Nagai T. et al. Transport of nerve growth factor encapsulated into liposomes across the blood-brain barrier: in vitro and in vivo studies. Journal of Controlled Release. 2005;105:106-19
45. Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cellular and Molecular Neurobiology. 2000;20:131-47
46. Habgood MD, Begley DJ, Abbott NJ. Determinants of passive drug entry into the central nervous system. Cellular and Molecular Neurobiology. 2000;20:231-53
47. Summerfield SG, Zhang Y, Liu H. Examining the uptake of central nervous system drugs and candidates across the blood-brain barrier. The Journal of Pharmacology and Experimental Therapeutics. 2016;358:294-305
Author contact
![]() Corresponding author: eva.jakabtothfr; khuloud.al-jamalac.uk
Corresponding author: eva.jakabtothfr; khuloud.al-jamalac.uk