- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(2):178-185. doi:10.7150/ntno.18720 This issue Cite
Research Paper
Gold Nanorod-based Photo-PCR System for One-Step, Rapid Detection of Bacteria
Jinjoo Kim1, 3*, Hansol Kim2, 4*, Ji Ho Park2, 4 ![]() , Sangyong Jon1, 3
, Sangyong Jon1, 3 ![]()
1. Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea;
2. Department of Bio & Brain Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea;
3. KAIST Institute for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea;
4. KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea.
* These authors contributed equally to this work.
Received 2016-12-12; Accepted 2017-3-24; Published 2017-5-6
Abstract

The polymerase chain reaction (PCR) has been an essential tool for diagnosis of infectious diseases, but conventional PCR still has some limitations with respect to applications to point-of-care (POC) diagnostic systems that require rapid detection and miniaturization. Here we report a light-based PCR method, termed as photo-PCR, which enables rapid detection of bacteria in a single step. In the photo-PCR system, poly(enthylene glycol)-modified gold nanorods (PEG-GNRs), used as a heat generator, are added into the PCR mixture, which is subsequently periodically irradiated with a 808-nm laser to create thermal cycling. Photo-PCR was able to significantly reduce overall thermal cycling time by integrating bacterial cell lysis and DNA amplification into a single step. Furthermore, when combined with KAPA2G fast polymerase and cooling system, the entire process of bacterial genomic DNA extraction and amplification was further shortened, highlighting the potential of photo-PCR for use in a portable, POC diagnostic system.
Keywords: bacterial detection, diagnosis, gold nanorod, polymerase chain reaction (PCR), photothermal heating.
Introduction
Numerous bacterial and viral infections pose severe threats to human health, placing urgent demands on the ability to rapidly and accurately diagnose them.[1] Of the various in vitro diagnostic tools available, the polymerase chain reaction (PCR) has been the method of choice for diagnosing infections owing to its accuracy and sensitivity. Despite these advantages, some drawbacks remain, including the relatively time-consuming thermal cycling steps and difficulty in miniaturizing the entire system, which limit its use in point-of-care (POC) diagnostics in the home or small clinic.[2-5] With conventional PCR, which utilizes a Peltier-based thermal cycler to generate the defining thermal cycles, heating and cooling of the bulk PCR mixture, rather than DNA amplification, is the time-consuming step, reflecting the relatively slow heat transfer between heating blocks and plastic PCR tubes.[6] Moreover, because the heat-conductivity of plastics is much lower than that of aqueous solutions, conventional PCR heating methods are not appropriate for achieving rapid thermal cycling. A more efficient solution would be to include a well-dispersed heating source in the PCR container itself. An example of this approach is nanoparticle-assisted PCR (nanoPCR), in which simple addition of gold nanoparticles (GNPs) to a conventional PCR enhances PCR efficiency and shortens the time to completion compared with a conventional PCR system.[7-11] The improvement provided by this nanoPCR system is attributable to the high heat conductivity of GNPs.[12-14] In addition, it has been shown that gold-based materials such as GNPs and gold thin films can be used as photothermal sources to achieve rapid thermal cycling for DNA amplification upon laser irradiation.[15-17] However, such approaches still require a separate cell lysis step to obtain genomic DNA prior to PCR thermocycling.[18, 19] A potential solution to this limitation is provided by recently reported cell lysis methods based on the laser-induced photothermal effect of GNPs or gold nanorods (GNRs), which is capable of lysing bacterial cells through heat shock, thereby releasing genomic DNA for subsequent conventional PCR.[20, 21] Therefore, integration of these two separate steps into a single test tube should enable rapid PCR-based detection.
Here, we present a novel photo-PCR system that can directly and rapidly amplify bacterial genomic DNA from intact bacterial cells. This photo-PCR system is particularly designed to accomplish rapid DNA amplification via thermal cycling through two temperature extremes (Figure 1A). We utilize poly(ethylene glycol)-modified gold nanorods (PEG-GNRs) as a photothermal heating source for both cell lysis and thermal cycling in the photo-PCR system because the GNR possess superior near-infrared (NIR) absorption and photothermal conversion properties than other gold-based nanomateirals [22, 23]. By adding PEG-GNRs to a PCR mixture and periodically switching on and off an 808-nm laser facing the solution, we show rapid and homogeneous control of the overall solution temperature, enabling heat lysis for DNA extraction and rapid thermal cycling between two temperatures for PCR. In this study we suggest that this photo-PCR system can be utilized in POC diagnostic tools for fast, one-step bacterial cell detection without the need of complex fabrication of PCR platforms.
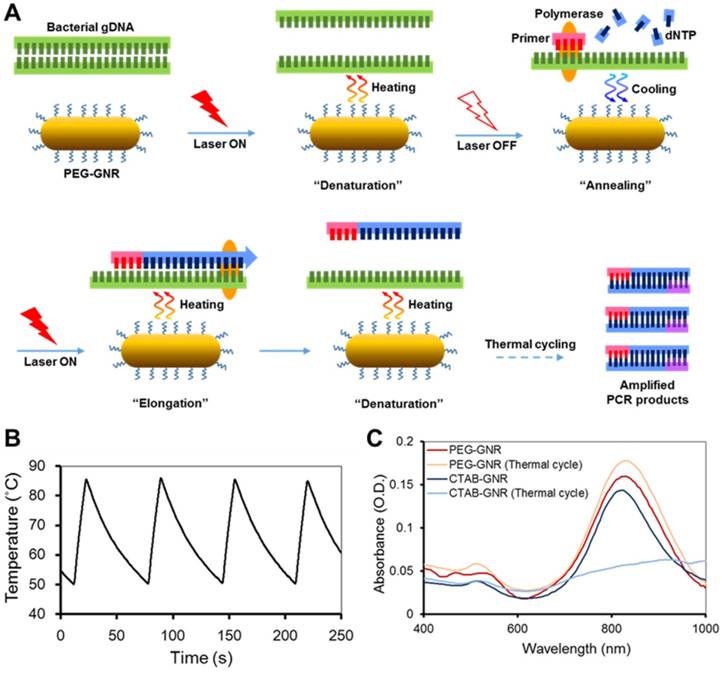
Characteristics of GNR-based photo-PCR. (A) Schematic depiction of bacterial genomic DNA (gDNA) amplification by photo-PCR. (B) Thermal cycling of photo-PCR. Representative temperature profile of 50 °C to 85 °C cycling with 0.24 nM PEG-GNRs. (C) UV/Vis absorption spectra of CTAB-GNRs and PEG-GNRs before and after thermal cycling with PCR mixture. Thermal cycling was performed under optimal conditions of 80 °C to 85 °C pre-heating for 3 min and 30 cycles of 50 °C to 85 °C using S. aureus cells. The samples were incubated with PCR mixture for 1 h prior to thermal cycling.
Methods
Preparation of PEG-GNRs
PEG-GNRs were prepared from cetyltrimethylammonium bromide (CTAB)-stabilized GNRs by exchanging CTAB with mPEG-thiol. The CTAB-stabilized GNRs with an aspect ratio of 4 were synthesized using a seed-mediated growth approach, as previously reported.[24] Briefly, 5 mL of 0.5 mM HAuCl4 (Sigma, St. Louis, USA) and 5 mL of 0.2 M CTAB (Sigma) solutions were prepared and mixed. Then, 600 μL of ice-cold 0.01 M NaBH4 (Sigma) solution was added to the mixture and sonicated for 4 min at 28 °C, after which the seed solution was allowed to stand at room temperature for 2 h. Growth solution was prepared by sequentially adding 5 mL of 0.001 M HAuCl4, 250 μL of 0.004 M AgNO3 (Sigma) and 70 μL of 0.0788 M ascorbic acid (Sigma) to 5 mL of 0.2 M CTAB solution. After gentle mixing, 12 μL of seed solution was mixed with growth solution at room temperature to initiate growth. When the longitudinal absorption peak reached ~800-820 nm, GNRs were centrifuged at 12,000 rpm for 15 min. The supernatant was discarded, and GNRs were re-suspended in distilled water. CTAB was exchanged with mPEG-thiol (2 kDa; Laysan Bio, Arab, USA) by adding 20 mg of mPEG-thiol to 10 mL of 3 optical density (O.D.). [25] GNRs were vortexed vigorously for 1 h, after which unincorporated mPEG-thiol was removed by centrifuging three times at 10,000 rpm for 10 min each.
Characterization of PEG-GNRs
Optical spectra were recorded using an UV/Vis spectrophotometer (SpectraMax Plus 384, Molecular Devices, Sunnyvale, USA), and zeta-potentials of GNRs were measured using dynamic light scattering (DLS; Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). Morphology and size distribution of PEG-GNRs were examined with a transmission electron microscope (TEM) (200 kV, JEOL Ltd., Tokyo, Japan)
Bacteria culture and genomic DNA extraction
Three types of bacteria, S. aureus (ATCC 25923), S. epidermidis (ATCC 12228) and E. coli (ER 2738), were cultured in Luria-Bertani broth. Bacterial genomic DNA was extracted using AccuPrep Genomic DNA Extraction Kits (Bioneer, Daejeon, Republic of Korea) according to the manufacturer's protocol.
Preparation of PCR reagents
Each PCR sample contained 5 μL of 10x Taq polymerase buffer, 0.8 μM each primer, 0.2 mM each dNTP, 2.5 U of Taq DNA polymerase (Beams Biotechnology, Seongnam, Republic of Korea), 5 μL of extracted genomic DNA or bacterial cells, and 5 μL of PEG-GNRs. The total volume of each PCR sample was brought up to 50 μL with PCR-grade water. For rapid photo-PCR samples, KAPA2G Fast PCR enzyme (KAPA2G Fast PCR kit; KAPA Biosystems, Wilmington, USA) was selected owing to its significantly faster extension rate compared with Taq polymerase. Each rapid PCR sample contained 2 μL of 5x KAPA2G Buffer A, 0.8 μM each primer, 0.2 mM each dNTP, 0.5 U of KAPA2G Fast DNA Polymerase, 1 μL of bacterial cells, and 2 μL of PEG-GNRs. The total volume of each PCR sample was brought up to 10 μL with PCR-grade water. S. aureus and S. epidermidis were detected using the primers 5'-GTA GTT AGC CGT GGC TTT CT-3' (forward) and 5'-GGG AAG AAC ATA TGT GTA AGT A-3' (reverse), and E. coli was detected using the primers 5'-GGA GTT AGC CGG TGC TTC T-3' (forward) and 5'-GGA GGA AGG GAG TAA AGT TAA T-3' (reverse), all obtained from Bioneer (Daejeon, Republic of Korea).[26]
Photo-PCR procedures
After preparing the 50-μL PCR mixture in a PCR tube as described above, 10 μL of mineral oil (Samchun Chemical, Seoul, Republic of Korea) was placed on top of the mixture to prevent evaporation, after which the PCR tube was irradiated using a benchtop fiber-delivered 808-nm laser system (5.6 W/cm2 , B&W TEK, Newark, USA). During irradiation, the surface temperature of the PCR solution was monitored with a thermographic infrared camera (FLIR, Wilsonville, USA). For rapid PCR, the PCR mixture (10 μL) was overlaid with 3 μL of mineral oil and a fan installed 10 cm away from the PCR tube was turned on during thermal cycle cooling periods.
Conventional PCR procedures
For conventional PCR controls, samples were amplified using a MJ mini-thermal cycler (Bio-Rad, Hercules, USA). For Taq polymerase samples, the thermal cycling profile consisted of 2 min at 95 °C, followed by 30 cycles of 95 °C for 15 s, 50 °C for 15 s and 72 °C for 15 s, with a final 5-min extension step at 72 °C. For KAPA2G Fast DNA polymerase samples, thermal cycling conditions were 1 min at 95 °C, followed by 30 cycles of 95 °C for 10 s, 50 °C for 10 s and 72 °C for 1 s, followed by a 30-s extension step at 72 °C.
Gel electrophoresis
PCR products were analyzed on 1.5% agarose gels using a Mupid-2plus electrophoresis apparatus (Optima Inc., Tokyo, Japan), with SYBR Safe DNA gel staining (Invitrogen, Carlsbad, USA). A 1-Kbp plus 100-bp DNA ladder (ELPIS-BIOTEC. Inc., Daejeon, Republic of Korea) was used to confirm product size. Gels were imaged using a Gel Doc XR+ imaging system (Bio-Rad), and band densities of individual samples were compared using GelQuant NET software provided by biochemlabsolutions.com.
Results and Discussion
Characterization of PEG-GNRs and NIR-induced thermal cycling
GNRs with an aspect ratio of 4, well known for their plasmonic photothermal effects in the NIR wavelength region, were chosen as a heating source for photo-PCR.[27-29] The surface of GNRs was modified with PEG molecules to minimize electrostatic interactions between GNRs and PCR reagents and improve the aqueous dispersity.[30, 31] PEG-GNRs were prepared by seed-mediated growth followed by exchange of CTAB with mPEG-thiol, as described in Methods. The zeta potential of PEG-GNRs (-5.8 ± 0.7 mV) was decreased from that of CTAB-GNRs (47.7 ± 0.2 mV), indicating the surface coverage of PEG. Localized surface plasmon resonance (LSPR) spectrum and TEM image of PEG-GNRs are shown in Figure S1. The spectrum showed typical characteristics of the GNRs, with a transverse LSPR peak at 515 nm and a longitudinal peak at 820 nm. We next utilized NIR photothermal capability of PEG-GNRs to perform thermal cycling of a PCR solution in the photo-PCR (Figure 1A). GNR-mediated photothermal heating enabled rapid thermal cycles of an aqueous solution from 50 °C to 85 °C (Figure 1B). PEG-GNRs dispersed well in the solution were reacted efficiently with incident light, thus leading to rapid temperature increase over the surrounding solution via nanoplasmonic photothermal heating.[23] The thermal stability of PEG-GNRs was evaluated by monitoring changes in LSPR spectra and TEM images. PEG-GNRs did not exhibit appreciable structural changes while maintaining their photo-stability even after 60 thermal cycles between 50 °C to 85 °C in pure water (Figure S2). Furthermore, no spectral abnormality was observed with PEG-GNRs in PCR mixture after 30 thermal cycles (Figure. 1C). These results indicate that PEG-GNRs can be used as a photothermal heating source to perform rapid and stable thermal cycling in subsequent PCR experiments.
Bacterial genomic DNA amplification using photo-PCR
We evaluated the DNA amplification ability of a GNR-based photo-PCR system compared with that of a conventional PCR system (Figure 1A). The optimal condition for photo-PCR was determined by varying several factors, including temperature, PEG-GNR concentration, and pre-heating condition. S. aureus genomic DNA was used as a target for these optimization experiments. The lower end temperature of the thermal cycle was set to 50 °C, based on the annealing temperature of primers, and the maximum temperature was varied from 80 °C to 95 °C at 5 °C intervals; each thermal cycle was repeated 30 times for all conditions. PCR products obtained using thermal cycles in the temperature ranges from 50 °C to 80 °C (50↔80 °C) or 50 °C to 85 °C (50↔85 °C) showed bands with the proper size (69 bp) on agarose gels. However, the band obtained using a thermal cycle in the temperature range from 50 °C to 90 °C (50↔90 °C) was faint, and no band was detected using a 50 °C to 95 °C (50↔95 °C) thermal cycling (Figure 2A), indicating that the thermal cycling with the upper limit temperature higher than 90 °C reduces the efficiency of photo-PCR. In photo-PCR, the thermal cycling is manipulated based on real-time measurement of the surface temperature of PCR solution. However, the actual temperature on the surface of GNRs during photothermal heating is significantly higher than that of PCR solution measured by thermographic infrared camera.[23] This surface-localized strong heating could yield low PCR efficiency due to inactivation of Taq polymerases at higher temperatures (half-lives: ~45-50 min at 95 °C and ~9 min at 97.5 °C).[32] This interpretation is also supported by the observation that a maximum temperature of 85 °C was sufficient to denature DNA in photo-PCR. Thus, the thermal cycling condition in the temperature range of 50↔85 °C was used in subsequent photo-PCR experiments.
Gel electrophoresis showing amplicons from photo-PCR and conventional PCR. (A) Effect of cycling temperature on photo-PCR using S. aureus genomic DNA (gDNA). (B) Effect of PEG-GNR concentration on photo-PCR using S. aureus gDNA. (C) Photo-PCR using E. coli and S. epidermidis gDNA.
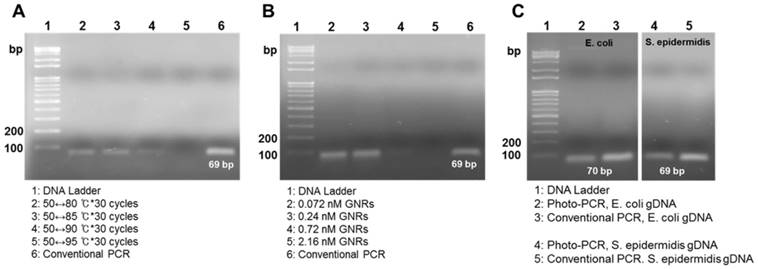
Previous nanoPCR-related research has reported that the concentration of heat-conductive GNPs is crucial for enhancing PCR efficiency, such that the GNP addition at the concentration greater than 1.0 nM was not beneficial in the PCR.[10] Therefore, we evaluated PCR efficacy as a function of PEG-GNR concentration from 0.072 nM to 2.16 nM. PEG-GNR concentrations of 0.07 nM and 0.24 nM were ideal for efficient photo-PCR (Figure 2B). By contrast, photo-PCR with higher concentrations of PEG-GNRs resulted in poor yields of PCR products although it increased both heating and cooling rates (Figure 2B, Figure S3 and Table S1), presumably due to spatial interference with DNA amplification and high probability for inactivation of Taq polymerases mediated by the overheated surface of GNRs. Therefore, we found that a PEG-GNR concentration of 0.24 nM was optimal for photo-PCR based on DNA amplification efficacy and total thermal cycling time.
As is the case for conventional PCR, pre-heating of samples at the denaturation temperature may enhance the yield of photo-PCR products by facilitating complete denaturation of target DNAs. We thus tested various pre-heating conditions (80↔85 °C, 85↔90 °C and 90↔95 °C for 30 s each) prior to the thermal cycling for photo-PCR. The amplicon bands obtained using 80↔85 °C and 85↔90 °C pre-heating conditions were slightly stronger than that obtained without pre-heating whereas the band for 90↔95 °C pre-heated sample was relatively faint compared with other samples (Figure S4). Importantly, the pre-heating sustained only for 30 s above 90 °C influenced the DNA amplification. These results re-confirm the notion that GNR-mediated photothermal heating of the PCR solution to above 90 °C can yield poor photo-PCR efficiency due to heat-mediated inactivation of Taq polymerases. Thus, the pre-heating condition of 80↔85 °C was chosen for subsequent photo-PCR experiments.
To assess generalizability, we next applied the photo-PCR conditions optimized with S. aureus genomic DNA to amplification of E. coli and S. epidermidis genomic DNAs. The photo-PCR was conducted in the PCR mixture containing 0.24 nM PEG-GNRs using a 80↔85 °C pre-heating step for 30 sec, followed by 30 thermal cycles of 50↔85 °C. The optimized photo-PCR conditions yielded appropriate-size amplicons for both target DNAs, which were similar to those produced by conventional PCR (Figure 2C). These experiments demonstrate that the reaction conditions optimized in this study can be utilized to amplify various bacterial genomic DNAs through photo-PCR.
We next examined whether photo-PCR system can also work for a much longer amplicon. When human β-actin gene with 318 bp was amplified under the thermal cycling between two temperatures (50 ℃↔ 85 ℃), the efficiency of photo-PCR was much lower than that of conventional PCR (Figure S5). However, when the elongation time for photo-PCR further increased with additional cycles (4 times of 68 ℃↔72 ℃), the amplification efficiency became significantly enhanced to be comparable to that of conventional PCR. As the amplicons smaller than 100 bp have been successfully used for bacterial detection,[26] we expect that the present photo-PCR enabling efficient detection of small-sized amplicons may be also suitable for the purpose.
One-step DNA extraction and amplification of bacterial cells using photo-PCR
It has been previously established that the photothermal effects of gold-based nanomaterials can be exploited to lyse bacterial cells and extract DNA.[20, 21] Hence, we took advantage of the pre-heating step to further extract genomic DNA from bacterial cells prior to DNA amplification in photo-PCR (Figure 3A). For one-step photo-PCR, bacterial cells (~104 colony forming unit (CFU) of S. aureus/50 μL), PCR reagents, and PEG-GNRs (0.24 nM) were mixed in a single tube. To achieve both DNA extraction and denaturation from bacterial cells, we extended the pre-heating time up to 180 s with the same temperature range (80↔85 °C). The DNA amplification was likely to increase with the pre-heating time (Figure 3B). The photo-PCR of bacterial cells with pre-heating for 180 s followed by thermal cycling resulted in the PCR efficacy comparable to that achieved in one-step conventional PCR in which genomic DNAs were extracted directly from bacterial cells during pre-heating. Thus, the 3-min pre-heating condition was used for all subsequent one-step photo-PCR of bacterial cells. These results demonstrate that bacterial cell lysis and DNA amplification can be done in one step through photo-PCR by simply manipulating the laser without the need for additional reagents or procedures.
Gel electrophoresis showing amplicons from one-step photo-PCR of bacterial cells. (A) Schematic depiction of one-step DNA extraction and photo-PCR of bacterial cells. (B) Effect of pre-heating time on one-step photo-PCR using S. aureus cells. Gel electrophoresis results demonstrate the dependence of yield on pre-heating time. (C) Representative temperature profile of the complete reaction. (D) One-step DNA extraction and photo-PCR with E. coli and S. epidermidis cells. Gel electrophoresis results show the amplicons obtained from samples containing E. coli or S. epidermidis.
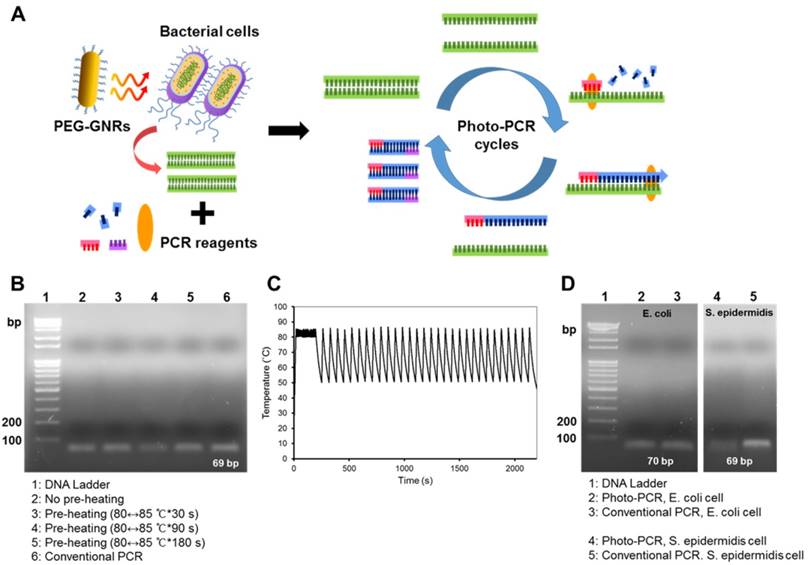
Other bacterial strains were also tested to confirm the effectiveness of one-step photo-PCR. Bacterial cell lysis and DNA amplification were conducted in one step with the optimized photo-PCR conditions including pre-heating step (80↔85 °C for 3 min) and subsequent thermal cycling (30 cycles of 50↔85 °C) (Figure 3C). Gel electrophoresis results showed that genomic DNAs of both Gram-positive (S. epidermidis) and Gram-negative (E. coli) bacteria were clearly detected after one-step photo-PCR (Figure 3D), indicating that GNR-mediated photothermal heating was successfully used to lyse bacterial cells and amplify DNA. Importantly, one-step photo-PCR reduced an overall reaction time to 38 min when compared with conventional PCR that typically requires longer reaction times (> 1 h) to complete DNA amplification.
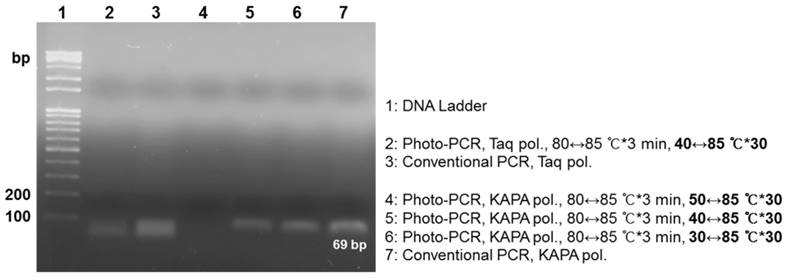
Lastly, we investigated how to reduce the total reaction time further for rapid one-step photo-PCR. We first introduced a cooling system to shorten the cooling cycle time because the cooling cycle that relies on natural cooling comprises 72% of the total thermal cycling time while the photothermal heating cycle constitutes only 12%. For the cooling system, we turned on the fan as the laser was turned off, except during the pre-heating step. We also decreased the volume of PCR mixture from 50 μL to 10 μL, and used KAPA2G Fast PCR polymerase instead of Taq polymerase due to its faster extension rate that may fit the accelerated cooling rate. We tested various thermal cycling conditions in this setup because rapid surface cooling of the low-volume solution with an external fan could cause a large discrepancy between the measured temperature and the solution temperature, particularly during the annealing. Gel electrophoresis results exhibited that new PCR samples successfully amplified appropriate-size PCR products under photo-PCR cycling conditions of 30 °C↔85 °C and 40 °C↔85 °C while they failed under 50 °C↔85 °C thermal cycling (Figure S6 and Figure 4). These results suggest that the actual temperature of the solution did not reach the annealing temperature under the 50 °C↔85 °C thermal cycling, resulting in failure of primers to anneal. Although the sample amplified under the 30↔85 °C thermal cycling condition showed slightly higher PCR efficiency compared with the 40↔85 °C cycling sample, it required a longer total cycling time (Table 1). Therefore, the 40↔85 °C thermal cycling was chosen as the optimal condition for rapid DNA amplification in this setup. The PCR efficiency of the sample containing Taq polymerase was lower than that of the sample containing KAPA2G Fast PCR polymerase under the 40 °C↔85 °C thermal cycling conditions, probably owing to its lower extension rate (Figure 4). Furthermore, the sensitivity of photo-PCR was compared with conventional PCR. As shown in Figure S7, photo-PCR was able to detect as low as 102 CFU of S. aureus, which was comparable to the detection limit of conventional PCR. Collectively, these results suggest that the optimized photo-PCR conditions including low solution volume, KAPA2G Fast PCR polymerase, and cooling fan enabled one-step cell lysis and PCR in a total time of 19 min.
Cycling time profile for one-step, rapid photo-PCR.
| Thermal Cycle (°C) | Heating (s) | Cooling (s) | Total thermal cycle (min) |
|---|---|---|---|
| 50 ↔ 85 | 6.8 | 13.2 | 13.8 |
| 40 ↔ 85 | 7.2 | 22.6 | 19.0 |
| 30 ↔ 85 | 8.8 | 40.7 | 28.3 |
Rapid photo-PCR with Taq polymerase or KAPA2G Fast PCR polymerase using thermal cycles with different low-end temperatures. Gel electrophoresis results demonstrate the dependence of yield on the type of polymerase and low-end temperature.
Conclusions
In this study, we demonstrated a rapid one-step bacteria lysis and DNA amplification using a photo-PCR system controlled with GNR-mediated nanoplasmonic heating. The high heat conductivity and NIR-induced photothermal conversion efficiency of PEG-GNRs significantly reduced the time required for thermal cycling. After optimization of photo-PCR conditions including GNR concentration, thermal cycling and pre-heating, bacterial cell lysis and DNA amplification was performed in a single step by manipulating laser irradiation. Furthermore, the photo-PCR system combined with KAPA2G Fast PCR polymerase and cooling fan amplified target genomic DNA directly from intact bacterial cells in a total time less than 20 min, while maintaining sufficient yield for clear detection. Although the set-up of photo-PCR presented herein has not been arranged for miniaturization yet, the thermographic infrared camera and the benchtop laser can be replaced with a small-sized thermocouple and a laser diode, respectively, enabling integration of all the components in a POC diagnostic system. Overall, this efficient bacteria-detection method based on photo-PCR offers great potential for applications in portable POC devices.
Supplementary Material
Supplementary figures and tables.
Abbreviations
PCR: polymerase chain reaction; POC: point-of-care; nanoPCR: nanoparticle-assisted PCR; GNP: gold nanoparticle; GNR: gold nanorod; PEG-GNR: PEG-modified gold nanorod; CTAB: cetyltrimethylammonium bromide; LSPR: localized surface plasmon resonance.
Acknowledgements
This work was supported by Global Research Laboratory (Grant No. 2015045887) and Bio&Medical Technology Development Program (Grant No. NRF-2012M3A9C6050125) of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fauci AS. Emerging and re-emerging infectious diseases: Influenza as a prototype of the host-pathogen balancing act. Cell. 2006;124:665-70
2. Fredricks DN, Relman DA. Application of polymerase chain reaction to the diagnosis of infectious diseases. Clin Infect Dis. 1999;29:475-86
3. Yamamoto Y. PCR in diagnosis of infection: Detection of bacteria in cerebrospinal fluids. Clin Diagn Lab Immun. 2002;9:508-14
4. Khot PD, Fredricks DN. PCR-based diagnosis of human fungal infections. Expert Rev Anti-Infe. 2009;7:1201-21
5. Yang S, Rothman RE. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis. 2004;4:337-48
6. Wittwer CT, Fillmore GC, Garling DJ. Minimizing the Time Required for DNA Amplification by Efficient Heat-Transfer to Small Samples. Anal Biochem. 1990;186:328-31
7. Wang JK, Cheng YN, Zhang M, Zhao H, Lin P, Yi L. et al. Development of a nanoparticle-assisted PCR (nanoPCR) assay for detection of mink enteritis virus (MEV) and genetic characterization of the NS1 gene in four Chinese MEV strains. Bmc Vet Res. 2015:11
8. Lin Y, Li J, Yao J, Liang Y, Zhang J, Zhou QF. et al. Mechanism of gold nanoparticle induced simultaneously increased PCR efficiency and specificity. Chinese Sci Bull. 2013;58:4593-601
9. Lou XH, Zhang Y. Mechanism Studies on NanoPCR and Applications of Gold Nanoparticles in Genetic Analysis. Acs Appl Mater Inter. 2013;5:6276-84
10. Li HK, Huang JH, Lv JH, An HJ, Zhang XD, Zhang ZZ. et al. Nanoparticle PCR: Nanogold-assisted PCR with enhanced specificity. Angew Chem Int Edit. 2005;44:5100-3
11. Li M, Lin YC, Wu CC, Liu HS. Enhancing the efficiency of a PCR using gold nanoparticles. Nucleic Acids Res. 2005:33
12. Hu M, Hartland GV. Heat dissipation for Au particles in aqueous solution: Relaxation time versus size (vol 106B, pg 7030, 2002). J Phys Chem B. 2003;107:1284 -
13. Maity S, Wu WC, Xu C, Tracy JB, Gundogdu K, Bochinski JR. et al. Spatial temperature mapping within polymer nanocomposites undergoing ultrafast photothermal heating via gold nanorods. Nanoscale. 2014;6:15236-47
14. Nguyen SC, Zhang Q, Manthiram K, Ye XC, Lomont JP, Harris CB. et al. Study of Heat Transfer Dynamics from Gold Nanorods to the Environment via Time-Resolved Infrared Spectroscopy. Acs Nano. 2016;10:2144-51
15. Roche PJR, Beitel LK, Khan R, Lumbroso R, Najih M, Cheung MCK. et al. Demonstration of a plasmonic thermocycler for the amplification of human androgen receptor DNA. Analyst. 2012;137:4475-81
16. Son JH, Cho B, Hong S, Lee SH, Hoxha O, Haack AJ. et al. Ultrafast photonic PCR. Light-Sci Appl. 2015:4
17. Son JH, Hong SG, Haack AJ, Gustafson L, Song MS, Hoxha O. et al. Rapid Optical Cavity PCR. Adv Healthc Mater. 2016;5:167-74
18. Pal R, Yang M, Lin R, Johnson BN, Srivastava N, Razzacki SZ. et al. An integrated microfluidic device for influenza and other genetic analyses. Lab Chip. 2005;5:1024-32
19. Lee JG, Cheong KH, Huh N, Kim S, Choi JW, Ko C. Microchip-based one step DNA extraction and real-time PCR in one chamber for rapid pathogen identification. Lab Chip. 2006;6:886-95
20. Cheong KH, Yi DK, Lee JG, Park JM, Kim MJ, Edel JB. et al. Gold nanoparticles for one step DNA extraction and real-time PCR of pathogens in a single chamber. Lab Chip. 2008;8:810-3
21. Wan W, Yeow JTW. Integration of nanoparticle cell lysis and microchip PCR for one-step rapid detection of bacteria. Biomed Microdevices. 2012;14:337-46
22. Jain PK, Lee KS, El-Sayed IH, El-Sayed MA. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: Applications in biological imaging and biomedicine. J Phys Chem B. 2006;110:7238-48
23. Ekici O, Harrison RK, Durr NJ, Eversole DS, Lee M, Ben-Yakar A. Thermal analysis of gold nanorods heated with femtosecond laser pulses. J Phys D Appl Phys. 2008:41
24. Nikoobakht B, El-Sayed MA. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater. 2003;15:1957-62
25. Niidome T, Yamagata M, Okamoto Y, Akiyama Y, Takahashi H, Kawano T. et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114:343-7
26. Chung HJ, Castro CM, Im H, Lee H, Weissleder R. A magneto-DNA nanoparticle system for rapid detection and phenotyping of bacteria. Nat Nanotechnol. 2013;8:369-75
27. Huang X, El-Sayed MA. Gold nanoparticles: optical properties and implementations in cancer diagnosis and photothermal therapy. Journal of Advanced Research. 2010;1:13-28
28. Mackey MA, Ali MR, Austin LA, Near RD, El-Sayed MA. The most effective gold nanorod size for plasmonic photothermal therapy: theory and in vitro experiments. The Journal of Physical Chemistry B. 2014;118:1319-26
29. Huang XH, Jain PK, El-Sayed IH, El-Sayed MA. Plasmonic photothermal therapy (PPTT) using gold nanoparticles. Laser Med Sci. 2008;23:217-28
30. Liopo A, Conjusteau A, Tsyboulski D, Ermolinsky B, Kazansky A, Oraevsky A. Biocompatible gold nanorod conjugates for preclinical biomedical research. Journal of nanomedicine & nanotechnology. 2012
31. Liopo AV, Conjusteau A, Chumakova OV, Ermilov SA, Su R, Oraevsky AA. Highly Purified Biocompatible Gold Nanorods for Contrasted Optoacoustic Imaging of Small Animal Models. Nanosci Nanotech Let. 2012;4:681-6
32. Lawyer FC, Stoffel S, Saiki RK, Chang S-Y, Landre PA, Abramson RD. et al. High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5'to 3'exonuclease activity. Genome Res. 1993;2:275-87
Author contact
![]() Corresponding authors: jihoparkac.kr; syjonac.kr
Corresponding authors: jihoparkac.kr; syjonac.kr