- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussions
Conclusion
Supplementary Material
Acknowledgements
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(1):103-113. doi:10.7150/ntno.18303 This issue Cite
Research Paper
A Vascular Permeability Assay Using an In Vitro Human Microvessel Model Mimicking the Inflammatory Condition
Joris Pauty1, 2*, Ryo Usuba1*, Haruko Takahashi1, Junichi Suehiro3, Kanoko Fujisawa1, Kiichiro Yano4, Tomohiro Nishizawa4, Yukiko T. Matsunaga1, 2 ![]()
1. Center for International Research on Integrative Biomedical Systems (CIBiS), Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan;
2. LIMMS/CNRS-IIS (UMI 2820), Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan;
3. Department of Pharmacology and Toxicology, Kyorin University School of Medicine, 6-20-2, Shinkawa, Mitaka-shi, Tokyo 181-8611, Japan;
4. End-Organ Disease Laboratories, R&D Division, Daiichi Sankyo Co., Ltd., 1-2-58, Hiromachi, Shinagawa-ku, Tokyo, 140-8710, Japan.
* These authors contributed equally to the work.
Received 2016-11-11; Accepted 2016-12-25; Published 2017-3-1
Abstract

The vascular barrier is an important function of the endothelium and its dysfunction is involved in several diseases. The barrier function of the endothelial cell monolayer is governed by cell-cell, cell-extracellular matrix (cell-ECM) contacts, and inflammatory factors such as thrombin, histamine or vascular endothelial growth factor. Several in vivo and in vitro assays that measure the vascular permeability induced by these factors have been developed. However, they suffer limitations such as being challenging for assessing details of biological processes at a cellular level or lacking the architecture of a vessel, that raise the need for new methods. In vitro 3D model-based assays have thus been developed but assays for investigating compounds that protects the barrier function are lacking. Here we describe the development of an in vitro three-dimensional (3D) vascular endothelium model in which we can manipulate the endothelial barrier function and permeability to molecules, which have a molecular weight similar to human serum albumin, allowing to assess the protective effect of compounds. A microvessel was prepared by culturing human umbilical vein endothelial cells (HUVECs) within a collagen gel on a polydimethylsiloxane (PDMS) chip. Using fluorescein isothiocyanate (FITC)-conjugated dextran (70 kDa, FITC-dextran) and confocal fluorescence microscopy, we showed that the microvessel presented an effective barrier function. We were then able to induce the loss of this barrier function by treatment with the inflammatory factor thrombin. The loss of barrier function was quantified by the extravasation of FITC-dextran into collagen matrix. Furthermore, we were able to analyze the protective effect on the endothelial barrier function of the cyclic adenosine monophosphate (cAMP) analog, 8-pCPT-2'-O-Me-cAMP (also called 007). In an attempt to understand the effects of thrombin and 007 in our model, we analyzed the adherens junctions and cytoskeleton through immunostaining of the vascular endothelial cadherin and actin, respectively. Our assay method could be used to screen for compounds modulating the barrier function of endothelial cells, as well as investigating mechanistic aspects of barrier dysfunction.
Keywords: Human umbilical vein endothelial cells, In vitro 3D model, Endothelial barrier function, Microvessel, Thrombin, Vascular permeability.
Introduction
The human circulatory system is a complex network of blood vessels with diameters ranging from a micro-millimeter to a centimeter [1]. The vascular endothelium lining the inner part of blood vessels ensures the barrier function that maintains blood within the circulatory system. However, molecules need to cross this barrier. The vascular endothelium maintains a certain permeability which allows essential small molecules such as ions, nutrients and water, to flow toward and from tissues to ensure cell survival, tissue function and homeostasis. This vascular permeability is finely regulated, as in the case of an injury, when the endothelium must allow larger elements, such as the components of the immune system, to access the tissue and heal it. Inflammatory factors such as the vascular endothelial growth factor (VEGF), histamine or thrombin induce this transient and non-pathological increase of vascular permeability [2, 3]. On the other hand, several pathologies are associated with a prolonged and/or excessive vascular permeability, which can occur due to weakening of the endothelial barrier or dysregulation of the inflammatory factors [4]. For example, in the first phase of diabetic retinopathy, the blood-retinal barrier is impaired resulting in a constant permeability. Inflammation then makes the disease progress toward a more severe phase called proliferative diabetic retinopathy, during which abnormal angiogenesis takes place and results in the formation of highly permeable vessels [5]. This relation between dysregulated angiogenesis and hyperpermeable vessels is seen in other pathologies such as cancer [6-8]. It has led to the emergence of a new therapeutic strategy: “vascular normalization”, which aims to correct the abnormalities of vessels. Bringing the vessels to a more “normal” state can reduce disease progression or help to achieve increased response to other therapies [9-11].
Because vascular permeability is an essential characteristic of the vascular system, methods to study it have been of interest since many years [12-14]. The first methods developed to study and measure vascular permeability mainly rely on in vivo assays, such as the Miles assay [15] or the use of blood vessels isolated from the organism known as ex vivo assays [16]. Because these models can be challenging to handle, it led to the development of the in vitro model known as the Transwell assay which allows to assess the permeability of a monolayer of endothelial cells with ease [17]. But it lacks the three-dimensional (3D) structure of a blood vessel. More recently, methods based on electrical resistance have been developed, such as transendothelial electrical resistance (TEER) and electrical cell-substrate impedance sensor (ECIS) [18]. Although these electrical resistance-based methods can be used to study barrier integrity and morphological change of endothelial cells, they do not allow assessing whether molecules such as proteins actually cross through the endothelial barrier and do not measure the permeability per se. With the extensive progress of tissue engineering, more sophisticated in vitro vasculature models can be achieved. These models recapitulate the 3D structure of blood vessels and enable control of the cellular environment [1]. Models have been developed by several teams using primary human cells such as human dermal microvascular endothelial cells (HDMECs) [19, 20] and human umbilical vein endothelial cells (HUVECs) [21, 22]. In these models, vascular permeability has been assessed to demonstrate the effective barrier function of tissue-engineered vessels and induced permeability of a microvessel made of HDMEC in response to the inflammatory factors histamine and thrombin has been reported [19]. However, assays for investigating compounds that protect the barrier function, which could thus be used for drug development aiming for vascular normalization, are lacking.
In this study, we proposed to build a human microvessel-on-a-chip that enables the study of compounds that protect the endothelial barrier function. We succeeded in quantitative analysis of the loss of barrier function using the microvessel model by observing the leakage of fluorescent molecules, which have a molecular weight similar to human serum albumin, with confocal fluorescent microscopy. The microvessel made of HUVECs showed efficient barrier function that could be disrupted by thrombin thereby causing a strong permeability. This model also enabled study of the protective effect of the cyclic adenosine monophosphate (cAMP) analog, 8-(4-Chlorophenylthio)-2'-O-methyl-adenosine 3',5'-cyclic monophosphate monosodium hydrate (8-pCPT-2′-O-Me-cAMP, also named 007), on the endothelial barrier function.
Materials and Methods
Materials and reagents
Reagents for cell culture
HUVECs and EGM-2 endothelial cell growth medium-2 BulletKit were purchased from Lonza (Basel, Switzerland). 0.5 w/v % trypsin-5.3 mM ethylendiaminetetraacetic acid (EDTA)-4Na solution and 10X Dulbecco's-phosphate buffered saline (PBS) (-) (2 g/L KCl, 80 g/L NaCl, 2 g/L KH2PO4, 11.5 g/L Na2HPO4) were ordered from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). 5 mL polystyrene round-bottom tube with cell-strainer cap (12 x 77 mm style) and 0.5% Trypan blue stain solution were purchased from Corning Falcon (Corning, NY, USA) and Nacalai Tesque, Inc. (Kyoto,Japan), respectively.
Reagents for preparing a neutralized collagen solution
Cellmatrix Type I-A (3 mg/mL, pH 3, collagen derived from porcine tendons by acid extraction) was ordered from Nitta Gelatin Co., Ltd. (Osaka, Japan). Collagen buffer was prepared in accordance with the manufacture's protocol. Briefly, in 100 mL 0.05 N NaOH, 2.2 g NaHCO3 (262 mM final) and 4.77 g HEPES (20 mM final) were dissolved. NaOH and NaHCO3 were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan); HEPES and 10X Hank's, from Sigma-Aldrich Co., Ltd. (Saint Louis, MO, USA).
Reagents for preparing an endothelium
Acupuncture needles (No.02, 0.20 mm x 30 mm, J type) were purchased from Seirin Co., Ltd. (Shizuoka, Japan). Bovine serum albumin (BSA) and dextran from Leuconostoc spp. (Mr 450,000-650,000) were ordered from Sigma-Aldrich. 10X D-PBS (-) and fibronectin (1 mg/mL) solution were purchased from Wako and Biomedical Technologies, Inc. (Stoughton, MA, USA), respectively.
Reagents for permeability assay
Rhodamine-conjugated Ulex europaeus Agglutinin 1 (UEA-1, 2 mg/mL) was purchased from Vector Laboratories, Inc. (Burlingame, CA, USA). Fluorescein isothiocyanate-dextran (FITC-dextran, 70 kDa) and EDTA were purchased from Sigma-Aldrich. Thrombin was ordered from General Electric Healthcare (Chicago, IL, USA) and resuspended at 1000 U/mL in PBS. 8-(4-Chlorophenylthio)-2'-O-methyl-adenosine 3',5'-cyclic monophosphate monosodium hydrate (8-pCPT-2′-O-Me-cAMP, also named 007) purchased from Sigma-Aldrich was reconstituted at 10 mM in PBS.
Reagents for immunofluorescence
4% (w/w) paraformaldehyde in PBS and Triton-X-100 were purchased from Wako and Sigma-Aldrich, respectively. The primary antibody targeting VE-cadherin (Rabbit mAb, D87F2) came from Cell Signaling Technology, Inc. (Danvers, MA, USA). The Alexa Fluor 568 phalloidin, secondary antibody Alexa Fluor 488 goat anti-rabbit and Hoechst 33342 were all purchased from Life Technologies - Thermo Fisher Scientific, Co. (Waltham, MA, USA).
Cell culture
HUVECs were cultured at 37°C in a humidified atmosphere of 5% CO2. Cells were passaged when a 70-80% confluence was achieved by rinsing once with PBS and then incubated with 0.25% trypsin-EDTA solution for 3 min at 37°C, 5% CO2. Trypsinized cells were then harvested in EGM-2 and seeded again with a dilution 1:4 in 10 cm culture dishes. In order to reduce the number of passages and to use the cells at the same passage when preparing microvessels, HUVECs were thawed upon reception, amplified for two passages and frozen again. When making microvessels, cells were thawed, seeded in 6 cm cultures dishes and used when they reached 70-80% confluence. For cell counting, trypsinized cells in EGM-2 were passed through a cell strainer to dissociate cell aggregates; dead cells were then stained with Trypan Blue and live cells were counted using a hemocytometer.
Design and fabrication of a microvessel
Microchips
Polydimethylsiloxane (PDMS)-based microchips (25 mm × 25 mm × 5 mm: width × length × height) that include needle inserting channels (300 µm in diameter) at both side were prepared with slight modifications of the method previously reported by J. Tien and colleagues [19] and kindly gifted by Dai Nippon Printing Ltd. (Tokyo, Japan). Briefly, a convex shaped mold was prepared using a 3D printer. The surface of the convex mold was treated with a release agent. A mixture of PDMS and a curing agent (SILPOT 184; Dow Corning Toray Co., Ltd., Tokyo, Japan) at a ratio of 10:1 (w/w) was cast on the convex mold equipped with 300-µm-diameter microneedles and subsequently cured at 37°C overnight. A concave PDMS device was then obtained by peeling it off the convex mold. The PDMS device and a glass coverslip (thickness No.1 0.12~0.17 mm, Matsunami, (Osaka, Japan)) were treated with O2 plasma (PDC-32G; Harrick Plasma (Ithaca, NY, USA)) for 60 seconds, before being bonded at 150°C for 30 min. The PDMS device was then check for leaks with 70% ethanol (Kanto Chemical) for 30 min before sterilization by ultraviolet (UV) light exposure for one hour under a cell culture hood.
Fabrication of a microvessel
The PDMS chip was first treated with O2 plasma for one minute and then sterilized under UV lights for 5 min. Meantime, an acupuncture needle (200 µm in diameter) was incubated in a 1% BSA in PBS solution at room temperature for at least 5 min, dried and sterilized by UV light exposure. The neutralized collagen solution was extemporaneously prepared by mixing on ice Cellmatrix Type I-A collagen solution with 10X Hank's buffer and 10X Collagen Buffer, with a volume ratio of 8:1:1. After warming up the PDMS chip for 5 min in a cell culture incubator (37°C, 5% CO2), the iced-cold neutralized collagen solution was added in the three chambers - the two reservoirs and the microvessel chamber. Immediately, the BSA-coated acupuncture needle was inserted within the PDMS channels and the collagen was withdrawn from the two reservoirs. The PDMS chip with the collagen and the needle were incubated in a cell culture incubator for 90 min to allow the collagen to gel. Once the collagen had fully gelled, the needle was withdrawn leaving a channel within the collagen. To prevent leakage of the chip through the PDMS channels used to insert the needle, two short needles (300 µm in diameter, 8 mm long) were inserted in the right and left PDMS channels located between the reservoirs and the external edge of the device (Fig.1A). The inner side of the collagen channel was then coated with fibronectin: 2 µL of a 10 µg/mL fibronectin in PBS solution were introduced from each opening of the collagen channel, incubated for 20 min at 37°C and removed. HUVECs were harvested and resuspended in EGM-2 containing 3% of dextran at a density of 1x107 cells/mL. 20,000 cells were then added near each opening of the collagen channel. After checking for the cell entering the collagen channel, the chip was flipped and incubated for 10 min in a cell culture incubator. The chip was flipped back and incubated an additional 5 min. 1 mL of warm medium was then added and the chip was further incubated for 4 hours. Finally, the medium was changed and cell culture was maintained for three days at 37°C, 5% CO2 with additional medium renewal every day.
Fabrication of a microvessel model and concept of the permeability assay (A) Left: a picture of the microchip with a microvessel (red); middle: bright field image of a microvessel; right: immunofluorescence 3D image of a microvessel showing the lumen, the adherens junctions (VE-cadherin in green) and the nuclei (blue). scale bars: 100 µm. (B) Fluorescent permeability assay; left: 3D-modeled image of the microchip showing the insertion of FITC-dextran; upper image: FITC-dextran within a microvessel; lower image: method for quantifying the permeability (see text for details), colors were converted from green to color-map using the ZEN software for better visualizing the variation of intensity of the FITC-dextran's fluorescence, ROI: region of interest, scale bar: 500 µm, color-map scale: fluorescence intensity expressed in arbitrary units. (C) Concept of the permeability assay: the insertion of 70 kDa FITC-dextran into a microvessel allows to visualize its barrier function; adding thrombin induces the disruption of the barrier function resulting in leakage of FITC-dextran; treating the microvessel with 007 prevents the disruption of the barrier function by thrombin.
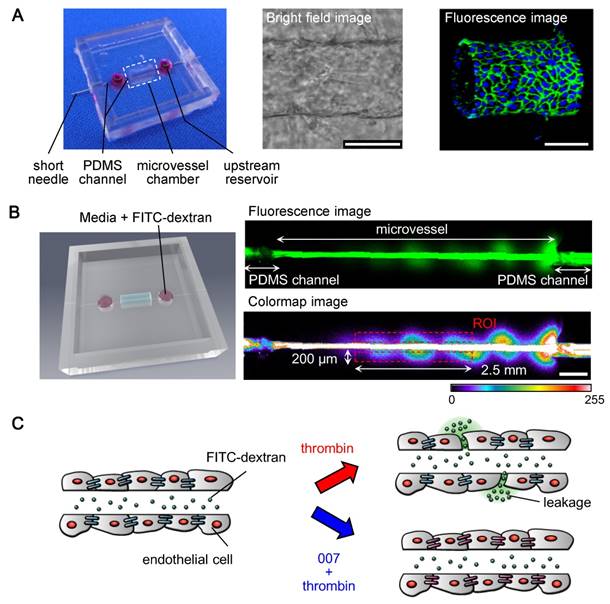
To measure the average diameter and length of a microvessel made with our method, bright field images of whole microvessels were taken 24 hours after cell seeding by using a Axiocam 506 mono Observer.Z1 microscope (Carl Zeiss Co., Ltd. (Oberkochen, Germany)) with a 20X lens. The length and diameter of a microvesel were estimated using the ZEN software (Carl Zeiss). The diameter of a microvessel was measured at the center of it.
Permeability assay
Three days after seeding the cells within the collagen channel, the microvessel was stained with 20 µg/mL rhodamine-conjugated UEA-1 in EGM-2 for 30 min to enable its visualization by confocal microscopy at a later stage of the assay. All further manipulations were performed by inserting the solutions in the right reservoir (upstream reservoir, Fig.1A and B) and all incubations were done at 37°C, 5% CO2. The microvessel was first treated with a 10 mM EDTA in EGM-2 solution for 30 min, followed by an incubation with a 100 U/mL thrombin in EGM-2 solution for 30 min. EDTA treatment was used to weaken the adherens junctions and thus help their opening by thrombin. To ensure that molecules of thrombin were properly distributed within the microvessel, the solution was removed from the left reservoir every 10 min and reinserted into the upstream reservoir. This handling was also done during the EDTA treatment as to expose the endothelial layer to the same stress. After the treatments and to visualize the permeability of the microvessel, the microchip was set up on a confocal microscope - Laser Scanning Microscope 700 (LSM700) (Carl Zeiss) in an incubation chamber that maintained a 37°C humidified atmosphere of 5% CO2 (INU Incubation System for Microscopes - model WSKM, Tokai Hit Co. (Fujinomiya, Japan)). Using a 10X lens, the rhodamine-conjugated UEA-1 staining was detected with the 555-nm-wavelength laser and used to focus the microscope. 15 µL of a 200 µg/mL FITC-dextran (70 kDa) in EGM-2 solution were then introduced (Fig.1B). Using the 488-nm-wavelength laser for detection and an optical section of 77.8 µm, images of the FITC's fluorescence were recorded every 2 min for a total of 10 min. Permeability was quantified by measuring the mean fluorescence intensity of the FITC-dextran detected within two regions of interest (“mean ROI”) outside a microvessel within the collagen. In details, for each microvessel, two regions of interest of 0.2 mm x 2.5 mm (0.5 mm2) were drawn using the ZEN software, aligned with the edges of the microvessel and centered on the middle of it (Fig.1B). The mean ROIs given by the ZEN software for the two boxes were added to give the fluorescence intensity that is used in the graphic representations.
To assess the protection of the barrier function realized by the cyclic adenosine monophosphate (cAMP) analog 007, the same method as described above was used with slight modifications; after incubation with EDTA and before treating with thrombin, the microvessel was incubated with 200 µM of 007 in EGM-2 for 30 min. For the quantitative study, the average fluorescence intensity and the standard deviation for six microvessels were calculated.
Immunofluorescence on a 2D model
Preparation of a 2D model
A 35-mm glass bottom culture dish (14-mm-diameter glass section) was treated with O2 plasma for one minute and sterilized under UV light for 5 min before adding 100 µL of ice-cold neutralized collagen type I-A solution to cover the glass section. The collagen gel layer was achieved by incubating the dish in a cell culture incubator (37°C, 5% CO2) for 30 min. The collagen layer was then coated with 100 µL of a 10 µg/mL fibronectin in PBS solution and incubated for 10 min. The fibronectin solution was removed and collagen gel was allowed to dry for 5 min at room temperature. 100 µL of a HUVEC suspension at 1x106 cells/mL was then added and incubated for 20 min to allow for cell attachment before adding gently 2 mL of warm EGM-2 medium. The growth medium was renewed 3 hours later. Cell culture was maintained for 3 days with medium renewal every day. The 2D model thus obtained was treated with EDTA and thrombin the same way as microvessels had been. Cells were then fixed by incubating for at least 15 min at room temperature with a 4% paraformaldehyde in PBS solution and kept at 4°C until immunofluorescence was performed.
Immunofluorescence on a 2D model
Following fixation and permeabilization (respectively 30 min in a 4% PFA in PBS solution and 5 min in a 0.5% TritonX-100 in PBS solution at room temperature), blocking was performed for overnight at 4°C with a 1% BSA in PBS solution (blocking solution). Then, incubation overnight at 4°C with the primary antibody anti-VE-cadherin diluted 1:200 in the blocking solution was done. Cells were washed with the blocking solution and incubated for 2 hours at room temperature with the secondary antibody (Alexa Fluor 488 goat anti-rabbit) and the Alexa 568 Phalloidin diluted in the blocking solution 1:500 and 1:200, respectively. After a wash with PBS, nuclei were stained by incubating the cells for 5 min at room temperature with Hoechst 33342 diluted 1:1000 in PBS. After several washes with PBS, cells were kept at 4°C until imaging.
Images were taken using a confocal microscope (LSM700, Carl Zeiss). Phalloidin, VE-cadherin and Hoechst 33342 were detected using 555-, 488- and 405-nm-wavelength lasers, respectively. Maximum intensity projection images were obtained with the ZEN software and used for the figure.
Results and Discussions
Fabrication of an in vitro microvessel with HUVECs
We designed a PDMS chip that included a rectangular chamber (2 mm x 6 mm x 3 mm: length x width x depth) at the center and two wells (3 mm in diameter and 3 mm in depth) framing and aligned with the central chamber (Fig. 1A). An empty channel (300 µm in diameter) crossed the whole chip through the rectangular chamber and the wells. The rectangular chamber, where a collagen bed was made and the vascular endothelium built, is referred to as the “microvessel chamber”. The wells at both sides enabled liquid and cell inputs and are called “reservoirs”. The empty channel allowed the insertion of a needle, used to create a tubular scaffold within the collagen for molding the endothelium.
A tubular scaffold within a collagen type I was prepared in the microvessel chamber using a 200-µm-diameter needle, as described in Materials and Methods. HUVECs were loaded into the scaffold by gently depositing them at both apertures of the collagen channel and allowing them to attach to the collagen gel. Following 24 hours of culture at 37°C with 5% CO2, the HUVECs had formed a tube showing quite consistent dimensions, for which we estimated the average length to be 5414 ± 141.2 µm for an average diameter of 157.4 ± 14.7 µm. Approximately 3 days after cell loading, the microvessel had homogenous VE-cadherin - a protein specific to the adherens junctions in the endothelium, and maintained a tubular structure with a lumen in its center, as highlighted by confocal microscopy (Fig. 1A).
Optimization of the experimental conditions to induce permeability with thrombin
To develop a stable and quantitative vascular permeability assay method realizing both thrombin-induced vascular permeability and its prevention by compounds (Fig.1B and C), it is important to identify the best treatment conditions for obtaining stimuli-responsive microvessels. Moreover, to measure the induced permeability, we chose to use a fluorescent microscopic method which consists in measuring the extravasated fluorescence of FITC-dextran with a molecular weight similar to human serum albumin, which is the most abundant protein in the human plasma. This fluorescence-based method requires to obtain a clear difference in leakage of FITC-dextran under the different treatments. We first validated the integrity of the barrier function in our model. We introduced a small volume (15 µL) of FITC-dextran (70 kDa), diluted in the culture medium EGM-2, from the upstream reservoir and monitored the fluorescence at 488 nm for 60 min using a confocal microscope (Fig. 2). No major leakage of FITC-dextran was observed, demonstrating that the microvessel presented an effective barrier function towards 70 kDa molecules. To induce the vascular permeability with thrombin, we first treated a microvessel with FITC-dextran in EGM-2 for 30 min in order to confirm its barrier function (Fig. 2). Then, we added 100 U/mL of thrombin in EGM-2 containing FITC-dextran and incubated for another 30 min. However, despite the high concentration of thrombin, no increase in permeability was observed. From these results, we conclude that our microvessels may have strong cell-cell junctions within the device.
Optimization of the method to induce vascular permeability of the microvessel model with thrombin (A) Left: timelines of the treatments applied to microvessels; right: representative images of the responses to the treatments (images taken at 60 min, scale bars: 500 µm, fluorescence intensity expressed in arbitrary units). (B) Representative curves of the time-dependent responses to the treatments: to optimize the time for treatments, we performed time-dependent experiments. FITC-dextran was added to each treatment solutions and images were taken every 2 min during the whole incubation time. Only fluorescence values measured every 10 min are shown (data of one representative experiment, experiments were performed two times for “untreated” and “thrombin” conditions, one time for “EDTA” condition and four times for “EDTA + thrombin” condition); the red arrow shows the time of thrombin addition.
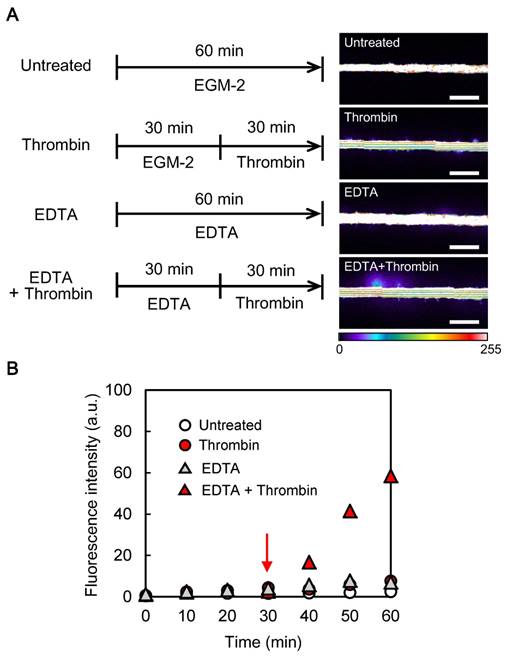
In order to make the microvessels responsive to thrombin, we hypothesized that the junctions should be weakened. It has been reported that thrombin induces endothelial permeability by causing a redistribution of the VE-cadherin and cell retraction [3]. From one hand, VE-cadherin is a calcium-dependent adhesion molecule, which is specific of endothelial adherens junctions. On the other hand, the effect of thrombin on cell contractility is caused by the remodeling of the cortical actin into actin stress fibers [23, 24]. Interestingly, it has been reported that this remodeling occurs rapidly, suggesting that the mechanical forces act early in the permeability induction [25, 26]. Based on this knowledge, we proposed that chelation of Ca2+ should weaken the adherens junctions by altering the VE-cadherin adhesion, facilitating their opening under the mechanical forces induced by thrombin signaling and the actin stress fibers. We therefore investigated the effect of chelating Ca2+ on the endothelial permeability by adding 10 mM EDTA in the EGM-2 containing FITC-dextran and monitored the fluorescence for 60 min. During the first 30 min, no leakage was observed (Fig. 2B, grey triangles). Thereafter, minor leakage started but did not evolve during the subsequent 30 min. This result indicates that exposing the HUVECs to EDTA for less than 60 min did not impair the barrier function of the endothelium.
Consistent to the previous result, we decided to employ a 30-min pre-treatment with EDTA before the addition of the thrombin (Fig. 2A). The assay was again performed using FITC-dextran in EGM-2, while monitoring of the fluorescence for 60 min. During pre-treatment with EDTA, no leakage was observed demonstrating the effective barrier function of the microvessel. Following the addition of thrombin, leakage of FITC-dextran rapidly occurred (Fig. 2B, red triangles), consistent with previously published works [3, 27]. Because EDTA alone did not induce leakage, and that thrombin could induce leakage only after EDTA treatment, it supported our hypothesis that EDTA acted by weakening the adherens junctions making them more susceptible to open under thrombin treatment.
Overall, these results demonstrate that our tissue-engineered microvessels presented an effective barrier function and that we could induce the dysfunction of this barrier with thrombin after pre-treatment with EDTA. We could also quantify the induced permeability by using confocal microscopy.
Assessing the protective effect of the cAMP analog 8-pCPT-2'-O-Me-cAMP (007) on the endothelial barrier function
As a proof-of-concept that our model could be used to assess the protective effect of compounds on the endothelial barrier function, we performed our assay with the cAMP analog 8-pCPT-2′-O-Me-cAMP (007), because it is known to decrease the endothelial permeability [28].
Considering the above results with EDTA treatment, all microvessels in these experiments were pretreated with EDTA for 30 min before use in the assay. To assess the effect of 007 in our model, we added to the method a 30-min incubation step with 200 µM 007 in EGM-2 between the pre-treatment with EDTA and the incubation with thrombin (Fig. 3A). We first confirmed that a 30-min incubation with EDTA followed by 60 min in EGM-2 did not affect our assay. Minor leakage occurred and did not increase markedly (Fig. 3). We then validated that the method still enabled the induction of endothelial permeability by thrombin.
Assessment of the protective effect of 007 on the endothelial barrier function (A) Left: timelines of the treatments applied to microvessels; right: representative images of the responses to the treatments (images taken at 90 min, scale bars: 500 µm, fluorescence intensity expressed in arbitrary units). (B) Representative curves of the time-dependent responses to the treatments: to optimize the time for treatments, we performed time-dependent experiments. FITC-dextran was added to each treatment solutions and images were taken every 2 min during the whole incubation time. Only fluorescence values measured every 10 min are shown (data of one representative experiment, experiments were performed one time for “untreated” condition, two times for “thrombin” and “007 + thrombin” conditions); the blue and red arrows show the addition of 007 and thrombin, respectively. (C) Quantitative study of the responses to the treatments: monitoring through time was not performed, the fluorescence intensity was only measured at the end of the method shown in A, i.e. 90 min after beginning of the experiment (error bars represent the standard deviations; n = 6).
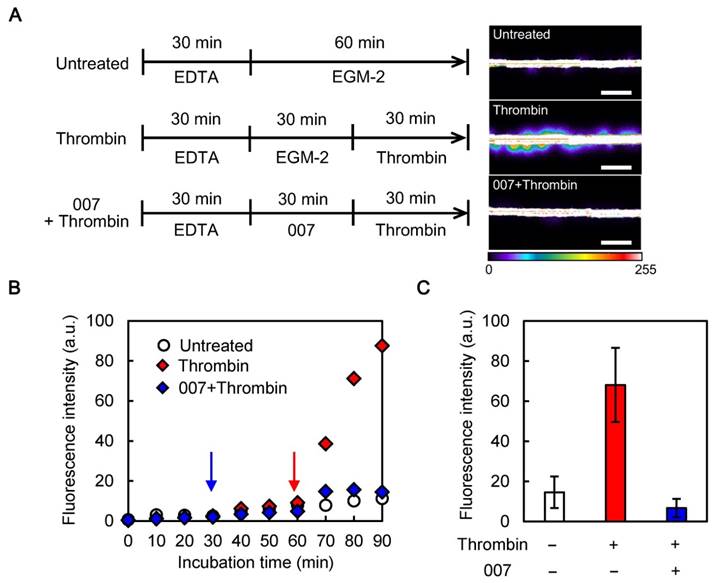
Because our permeability assay was still viable, we were able to assess the effect of 007. After the 30-min pre-treatment with EDTA, 30-min of incubation with 007 and 30-min of treatment with thrombin (Fig. 3A), some minor leakage could be seen but it did not increase markedly (Fig. 3B). A quantitative study showed that thrombin increased the leakage by about seven times (a fluorescence intensity increase from around 10 a.u. to 70 a.u.), while treatment with 007 blocked the increase (Fig. 3C). This result demonstrates that 007 successfully prevented the induction of the endothelial permeability by thrombin in our assay.
Immunofluorescence analysis of the effects of thrombin and 007 on adherens junctions and actin cytoskeleton
During inflammation, adherens junctions play a major role in the control of vascular permeability [29-32]. Adherens junctions also influence the cytoskeleton and are influenced by it. Furthermore, there are evidences that adherens junctions affect the organization of tight junctions. Therefore, to investigate the effects of thrombin and 007 in our method, we focused on adherens junctions and cytoskeleton. 007 has been described to tighten the adherens junctions and decrease the endothelial permeability by counteracting the effects of thrombin [28, 33, 34]. 007 induces a redistribution of the VE-cadherin - essential component of the adherens junctions, and promotes actin reorganization by two ways: it increases the circumferential actin and down-regulates the activation of Rho GTPases - one of the events triggered by thrombin that leads to actin stress fibers formation [23, 24]. Therefore, we analyzed the effect of 007 and thrombin on adherens junctions and cytoskeleton by immunostaining of VE-cadherin and actin fibers, respectively.
The immunofluorescence was performed on a 2D model that recapitulates the main features of our in vitro 3D model, i.e. a monolayer of HUVECs attached onto a matrix of collagen type I. The 2D model was treated in the same way as the in vitro 3D model. We used a 2D model because the detailed analysis of adherens junctions and cytoskeleton required to use a high magnification - typically 40X, on a confocal microscope. However, focusing on a microvessel with a 40X-lens revealed to be challenging with our microchip. Although we could sometime succeed, we were limited in the plan that we could reach and therefore, we preferred to rely on a 2D model for a more reproducible and convincing analysis.
First, we analyzed the effect of the EDTA treatment. Following 30-min of incubation with EGM-2 containing EDTA, we could see a broadening of the VE-cadherin signal at the cell-cell junctions and the loss of VE-cadherin in only few areas (Fig.4A). It indicated that the adherens junctions were globally maintained, although weakened. In addition, the actin cytoskeleton concentrated at the periphery of the cells colocalizing with the VE-cadherin and less filaments of actin were seen within the cytoplasm. Filaments of actin are known as stress fibers and correlate with an increased permeability, while cortical actin is associated with a strengthening of the cell-cell junctions [23, 25, 33]. These observations supported our hypothesis that chelating Ca2+ by EDTA would weaken the adherens junctions without significantly affecting the cytoskeleton. Interestingly, some stress fibers could also be seen in the control (EGM2). It is known that a cell monolayer is not static. Some areas are submitted to mechanical stress due to tensions exerted by other areas where cells contract, therefore, explaining the presence of actin stress fibers in the untreated condition. After EDTA pre-treatment, when the monolayer was subsequently exposed to EGM-2, no major changes were observed for the first 30 min (Fig.4B, left panels). However, after 60 min, VE-cadherin signal became broader and missing in some areas (Fig.4C, left panels). Furthermore, some actin stress fibers could be seen (Fig.4B and C, left panels). The focal absence of VE-cadherin, broad nature of adherens junctions and presence of actin stress fibers, explain the weak leakage observed in Fig.3 (white circles and bar).
Immunofluorescence on 2D model showing the effect of the treatments on the adherens junctions and actin cytoskeleton (A, B, C) Immunofluorescence of the VE-cadherin (VE-cad, green), actin cytoskeleton (red) and nucleus (blue) on the 2D-model treated the same way as the microvessel model as shown on the timeline in the upper panel (➤: missing VE-cadherin, →: actin stress fiber, scale bars: 20 µm). (A) Effect of 30-min pre-treatment with 10 mM EDTA. (B) Effect of 30-min incubation with EGM-2 or 200 µM 007 after EDTA. (C) Left: effect of 60-min incubation with EGM-2 after EDTA; middle and right: effect of 30-min treatment with 100 U/mL thrombin after EDTA and EGM-2 or 007.
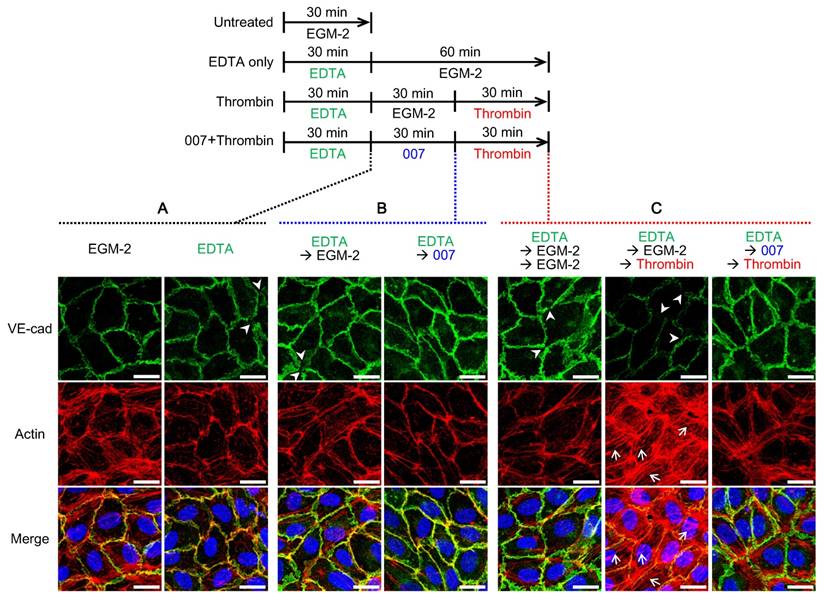
Then, we analyzed the effect of 007. When 007 was introduced after pre-treatment with EDTA, the VE-cadherin signal at the cell-cell boundary became smoother, suggesting a recovery of the adherens junctions. Regarding the actin cytoskeleton, the 007 treatment reduced the stress fibers in favor of the cortical actin implying a strengthening of the cell-cell junctions (Fig. 4B). These observations are in accordance with previous works [33, 35].
Finally, we investigated the effect of thrombin and assessed the effect of 007 in that context. When thrombin was added 30 min after EDTA and EGM-2 exposure, the VE-cadherin became missing in several areas (Fig. 4C, middle panel). Furthermore, the actin cytoskeleton strongly rearranged from cortical actin to stress fibers (Fig. 4C, middle panel compared to left panel). Interestingly, actin was missing in areas were VE-cadherin was missing as well. This result is in accordance with the known effects of thrombin [23, 24] and explains the successful induced-permeability observed in Fig. 3 (red squares and bar). When incubation with 007 was performed before the thrombin treatment, the VE-cadherin signal showed some broadness, but was maintained all around the cell. Actin fibers were formed, but they were mainly localized in the periphery of the cell. This suggested they were cortical bundled fibers rather than stress fibers (Fig.4C, right panels). The maintenance of the VE-cadherin at the junctions as well as the cortical actin explain the prevention of leakage observed in Fig. 3 (blue squares and bar).
For complementary analysis of the cell-cell junctions, we also assessed the tight junctions by immunostaining of the protein ZO-1 (Fig. S1). EDTA pre-treatment followed by incubation with EGM2, led to a thinner ZO-1 signal at the cell-cell boundaries compared to the initial signal (Fig. S1B compared to S1A). When the HUVECs were further treated with thrombin, the ZO-1 signal became broader suggesting that the tight junctions were stretched. Interestingly, the direction in which the ZO-1 signal was broader seemed to correlate with the direction of the actin stress fibers. When incubation with 007 was performed before the thrombin treatment, ZO-1 signal looked similar to that of the control condition, i.e. EDTA-pre-treatment only. These results support the previous ones and state that 007 prevents the loss of barrier function by strengthening the cell-cell junctions in our experimental conditions.
Conclusion
In this study, we reported the development of an in vitro 3D endothelial cell-based microvessel in which we are able to manipulate endothelial barrier function and permeability to assess the protective effect of compounds. Using a PDMS microchip, we created an in vitro microvessel of HUVECs showing mature cell-cell junctions and an effective barrier function. The microchip allowed us to perform confocal microscopy, with which we could monitor the endothelial extravasation of 70 kDa FITC-dextran into the collagen matrix. We identified conditions that allowed us to induce permeability in the microvessel in response to thrombin and could prevent the effect of thrombin and limit the endothelial permeability by using 007. On another hand, using a 2D model, we performed immunofluorescence in an attempt to understand the drug-response observed in our method. It revealed that structural changes of the endothelial cell junctions and cytoskeleton which occurred in our model, recapitulate the known effect of thrombin and 007.
Although the quantification method that we use, does not allow to precisely determine a permeability coefficient, it is easy to set-up as no complicated analytical model is required. In addition, the obtained values can be compared between different compounds in order to identify the most effective in preventing loss of barrier function. Therefore, this method can be easily used in initial screening of candidate compounds that are believed to reduce the endothelial permeability or strengthen the barrier function of endothelial cells. Such compounds are of interest in the development of new therapeutic strategies whereby limiting an excessive permeability would reduce the progression of a disease or increase the efficiency of other therapies, e.g. for cancer and retinopathy. We used thrombin to develop the method. However, we aim to extend this method to suit specific diseases, i.e. to reproduce pathological permeability using specific factors observed in any disease for which we wish to screen compounds.
Supplementary Material
Supplementary method and figure S1.
Acknowledgements
This work was partly supported by the Japan Society for the Promotion of Science (JSPS) Core-to-Core Program to Y.T.M., Shoreikai fellowships to Y.T.M and R.U. J.P is an International Research Fellow of the JSPS (P15767). The authors thank Mr. Yasuhiro Ookawa and Dr. Koji Fujimoto (Dai Nippon Printing Co., Ltd.) for their assistances in the PDMS device fabrication, Dr. Yosuke Hiraoka (Nitta Gelatin Inc.) for his kind support in collagen preparation, Mr. Isamu Matsuda (Waseda University) for his assistance in the making protocols for microvessel formation and permeability assay using a confocal microscope, Ms. Eri Otsuka (The University of Tokyo) for her technical assistance in the preparation of microvessels and Mr. Takashi Ando for his contribution to the artwork.
Abbreviations
3D: three-dimensional; BSA: bovine serum albumin; cAMP: cyclic adenosine monophosphate; ECM: extracellular matrix; EDTA: ethylendiaminetetraacetic acid; EGM-2: endothelial cell growth medium 2; FITC: fluorescein isothiocyanate; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HDMEC: human dermal microvascular blood cell; HUVEC: human umbilical vein endothelial cell; kDa: kilodalton; PBS: phosphate buffered saline; PDMS: polydimethylsiloxane; PFA: paraformaldehyde; ROI: region of interest; UEA-I: Ulex europaeus Agglutinin I; UV: ultraviolet; VE-Cad: vascular endothelial cadherin; VEGF: vascular endothelial growth factor.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bogorad MI, DeStefano J, Karlsson J, Wong AD, Gerecht S, Searson PC. Review: in vitro microvessel models. Lab Chip. 2015;15:4242-55
2. Claesson-Welsh L. Vascular permeability-the essentials. Ups J Med Sci. 2015;120:135-43
3. Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16:488-96
4. Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol. 2008;15:243-9
5. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012;366:1227-39
6. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-57
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
8. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273-86
9. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071-121
10. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417-27
11. Nakajima M, Cooney MJ, Tu AH, Chang KY, Cao J, Ando A. et al. Normalization of retinal vascular permeability in experimental diabetes with genistein. Invest Ophthalmol Vis Sci. 2001;42:2110-4
12. Bersini S, Yazdi IK, Talo G, Shin SR, Moretti M, Khademhosseini A. Cell-microenvironment interactions and architectures in microvascular systems. Biotechnol Adv. 2016;34:1113-30
13. Hasan A, Paul A, Vrana NE, Zhao X, Memic A, Hwang YS. et al. Microfluidic techniques for development of 3D vascularized tissue. Biomaterials. 2014;35:7308-25
14. Auger FA, Gibot L, Lacroix D. The pivotal role of vascularization in tissue engineering. Annu Rev Biomed Eng. 2013;15:177-200
15. Miles AA, Miles EM. Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea-pigs. J Physiol. 1952;118:228-57
16. Yuan Y, Chilian WM, Granger HJ, Zawieja DC. Permeability to albumin in isolated coronary venules. Am J Physiol. 1993;265:H543-52
17. Siflinger-Birnboim A, Del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111-7
18. Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci U S A. 1992;89:7919-23
19. Chrobak KM, Potter DR, Tien J. Formation of perfused, functional microvascular tubes in vitro. Microvasc Res. 2006;71:185-96
20. Wong KH, Truslow JG, Tien J. The role of cyclic AMP in normalizing the function of engineered human blood microvessels in microfluidic collagen gels. Biomaterials. 2010;31:4706-14
21. Baker BM, Trappmann B, Stapleton SC, Toro E, Chen CS. Microfluidics embedded within extracellular matrix to define vascular architectures and pattern diffusive gradients. Lab Chip. 2013;13:3246-52
22. Kim S, Lee H, Chung M, Jeon NL. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip. 2013;13:1489-500
23. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279-367
24. Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;2007:re8
25. Thurston G, Turner D. Thrombin-induced increase of F-actin in human umbilical vein endothelial cells. Microvasc Res. 1994;47:1-20
26. Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ. 2014;4:535-51
27. Draijer R, Atsma DE, van der Laarse A, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res. 1995;76:199-208
28. Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG. et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901-6
29. Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009;335:17-25
30. Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467-77
31. Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol. 2008;28:223-32
32. Gavard J. Endothelial permeability and VE-cadherin: a wacky comradeship. Cell Adh Migr. 2014;8:158-64
33. Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966-72
34. Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950-5
35. Noda K, Zhang J, Fukuhara S, Kunimoto S, Yoshimura M, Mochizuki N. Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through alpha- and beta-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol Biol Cell. 2010;21:584-96
Author contact
![]() Corresponding author: Yukiko T. Matsunaga, Ph.D., Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo Japan 153-8505; Tel: +81-3-5452-6470; Fax: +81-3-5452-6471 E-mail: matu-tokyo.ac.jp
Corresponding author: Yukiko T. Matsunaga, Ph.D., Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo Japan 153-8505; Tel: +81-3-5452-6470; Fax: +81-3-5452-6471 E-mail: matu-tokyo.ac.jp