- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(1):59-79. doi:10.7150/ntno.17896 This issue Cite
Research Paper
Novel Hyaluronic Acid Conjugates for Dual Nuclear Imaging and Therapy in CD44-Expressing Tumors in Mice In Vivo
Ravindra Dhar Dubey1, Rebecca Klippstein2*, Julie Tzu-Wen Wang2*, Naomi Hodgins2, Kuo-Ching Mei2, Jane Sosabowski3, Robert C. Hider2, Vincenzo Abbate2, Prem N. Gupta1, Khuloud T. Al-Jamal2 ![]()
1. Formulation & Drug Delivery Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu-180001, India.
2. Institute of Pharmaceutical Science, Faculty of Life Sciences & Medicine, King's College London, Franklin-Wilkins Building, London SE19NH, United Kingdom.
3. Centre for Molecular Oncology, Bart's Cancer Institute, Queen Mary University of London, London, EC1M 6BQ, United Kingdom.
*Contributed equally to the work.
Received 2016-10-16; Accepted 2016-11-28; Published 2017-1-1
Abstract

Hyaluronic acid, a natural CD44 receptor ligand, has attracted attention in the past years as a macromolecular delivery of anticancer agents to cancer. At the same time, the clinical applications of Gemcitabine (Gem) have been hindered by its short biological half-life, high dose and development of drug resistance. This work reports the synthesis of a hyaluronic acid (HA) conjugate for nuclear imaging, and in vivo Gem delivery to CD44-expressing solid tumors in mice. HA was individually conjugated, via amide coupling, to Gem (HA-Gem), 4'-(aminomethyl)fluorescein hydrochloride (HA-4'-AMF) or tris(hydroxypyridinone) amine (HA-THP) for cancer therapy, in vitro tracking or single photon emission computed tomography/computed tomography (SPECT/CT) imaging, respectively. Gem conjugation to HA was directly confirmed by nuclear magnetic resonance (1H NMR), gel permeation chromatography (GPC) and UV-visible spectrometry, or indirectly by a nucleoside transporter inhibition study. Gem conjugation to HA improved its plasma stability, reduced blood hemolysis and resulted in delayed cytotoxicity in vitro. Uptake inhibition studies in colon CT26 and pancreatic PANC-1 cells, by flow cytometry, revealed that uptake of fluorescent HA conjugate is CD44 receptor and macropinocytosis-dependent. Gamma scintigraphy and SPECT/CT imaging confirmed the relatively prolonged blood circulation profile and uptake in CT26 (1.5 % ID/gm) and PANC-1 (1 % ID/gm) subcutaneous tumors at 24 h after intravenous injection in mice. Four injections of HA-Gem at ~15 mg/kg, over a 28-day period, resulted in significant delay in CT26 tumor growth and prolonged mice survival compared to the free drug. This study reports for the first time dual nuclear imaging and drug delivery (Gem) of HA conjugates to solid tumors in mice. The conjugates show great potential in targeting, imaging and killing of CD44-over expressing cells in vivo. This work is likely to open new avenues for the application of HA-based macromolecules in the field of image-guided delivery in oncology.
Keywords: Gemcitabine, Hyaluronic acid, Image-Guided Delivery, SPECT, Nuclear Imaging, CD44-receptor, Endocytosis.
Introduction
Polymer-based nanocarriers, specially polymer-drug conjugates, have attracted much attention in cancer therapy [1] due to flexibility in macromolecular synthetic methods, diversity of polymers in terms of source, composition and ease of functionalization [2, 3]. Conjugation of low molecular weight drugs to polymers alters the drug's organ biodistribution profile, provides target specific delivery and reduces systemic toxicity. There are several polymer-anticancer drug conjugates that have reached clinical development [4-10] paclitaxel-polyglutamic acid conjugate (XYOTAXTM), for example, showed promising results in phase III clinical trials in women with non-small-cell lung cancer [11]. Despite these successes one key issue that is often neglected in the clinical application of macromolecular delivery is quantifying how much of the injected polymer reaches the tumor site. This is further complicated by the heterogeneity in Enhanced Permeation and Retention (EPR) and targeting receptors expression among the different tumors.
Positron Emission Tomography (PET) and single photon emission computed tomography/computed tomography (SPECT) are two of the main imaging methods used in clinical practice [12, 13]. Radionuclides used in PET imaging have very short half-lives, so finding an instant nuclear labelling method of high radiolabelling efficacy, i.e., no purification is needed, is always sought. Tris(hydroxypyridinone) (THP) has been reported to offer instant and stable chelation with gallium radiometals, offering an attractive labelling approach in Positron Emission Tomography (PET) (68Ga) and SPECT (67Ga) imaging of peptides [14, 15].
Gemcitabine (Gem) is a bifluorinated deoxycytidine analogue prodrug, which internalizes into cells via the human equilibrative nucleoside transporter-1 (hENT1), a sodium-independent pathway. Following its entry into cells, Gem is phosphorylated into its mono-, di- and tri-phosphate derivatives, dFdCMP, dFdCDP and dFdCTP, respectively, by the action of deoxycytidine kinase (dCK). The active metabolite dFdCTP incorporates into the DNA of cancer cells, as a false nucleoside moiety, leading to cell proliferation inhibition and cell apoptosis [16-18]. It demonstrates a broad range of anticancer activity in hematological malignancies and against several solid tumor types [19, 20].
Gemcitabine·HCl is marketed clinically, in more than 70 countries, as a parenteral formulation (Gemzar®), injected via intravenous (i.v.) infusion at a dose of 1000-1250 mg/m2 over 30 min, on a weekly basis, for 3-4 weeks [21]. Its clinical benefit, however, is limited by its short plasma half-life (about 15 min) [22]; it undergoes rapid deactivation by the action of deoxycytidine deaminase, present in the blood and liver [23], followed by renal excretion. Such pharmacokinetic (PK) and pharmacodynamic (PD) profiles suggest that more frequent dosing is required. In some cases, daily administrations or prolonged infusions are required so that a therapeutic effect of the drug can be obtained [24, 25]. Higher doses of Gem are associated with serious side effects such as myelosuppression, thrombocytopenia, mild and transient neutropenia and anemia [26]. In addition to the PK/PD complexity, nucleoside transporters are needed for intracellular internalization of Gem and so drug resistance develops over time, diminishing Gem therapeutic efficacy [20, 27].
Polymer-Gem conjugates have been proposed, as one of the solutions, to improve Gem's plasma stability, therapeutic efficacy and therapeutic index. Gem conjugation to polyethylene glycol (PEG) [28] and linoleic acid [29], via its N4-position, prolonged blood circulation time and improved its bioavailability as compared to the free drug. Other types of polymer conjugate such as an amphiphilic polymer like poly(ethyleneglycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate) [30] and the polyisoprenoy prodrug of Gem [31] have been proposed, reporting improvements in plasma stability and in vivo antitumor efficacy after i.v. injection, in a MIAPaCa-2 cell derived xenograft model. In another approach, Gem has been conjugated to fatty acids (C3-C16) and subsequently incorporated into liposomes [32, 33]. This approach improved plasma stability, enhanced in vitro cytotoxicity and in vivo antitumor efficacy of all conjugates, and particularly stearoyl-Gem, compared to unmodified Gem [34].
HA is a natural polysaccharide, composed of an alternating unit of N-acetyl-D-glucosamine and D-glucuronic acid, and is present in the extracellular matrix and synovial fluid [35]. It has been widely explored in anticancer drug delivery owing to its unique properties as a biodegradable, biocompatible, non-toxic, and non-immunogenic polymer. Targeting the CD44-receptor, over expressed in several types of cancer cells [36], attracted the attention of the scientific community, complemented by the experimental work showing improvements in the therapeutic efficacy of some anticancer drugs [37-39]. For example, paclitaxel-hyaluronan bioconjugate, targeting CD44 receptor, exhibited potent therapeutic activity against IGROV-1 and OVCAR-3 ovarian cancer xenografts model in vivo [40]. Multi-prodrug nanocarriers have been developed in which Gem and paclitaxel were coupled separately to HA and poly (L-lysine)-carboxylate, respectively. A synergistic anticancer activity was shown in HuCCT1 tumor bearing BALB/c nude mice following tail vein injection of both conjugates [41].
Considering the above drug shortcomings, we synthesized a Gem-HA conjugate, via an amide linkage as a more stable analogue in vivo. Conjugation of Gem to HA using the amide linkage has not been reported before. In general amide linkages are more stable than ester linkages so it is hypothesized that this strategy is likely to improve the plasma stability of Gem; prolong it's biological activity, and if successfully delivered to solid tumors, could delay tumor growth of CD44-expressing solid tumor models, following i.v. administration. The only study previously reporting Gem conjugation to HA utilized an ester linkage [41]. Moreover, it was reasoned that replacement of the Gem moiety with a fluorescent or nuclear imaging probe would allow in vitro (intracellular trafficking) or in vivo imaging (SPECT/CT), respectively. Conjugation of a THP chelator to HA has not been attempted previously, and if successfully achieved, could permit both PET and SPECT imaging.
In this study, a tripodal HA conjugate; enabling the targeting, imaging and therapy of CD44-expressing cancer cells in vivo was synthesized. Chemical characterization, in vitro cell tracking and cytotoxicity studies were performed in a range of cancer cells. In vivo tumor biodistribution studies were carried out in two CD44-expressing tumor-bearing mice; the highly vascularized murine colon cancer (CT26) and the highly resistant human pancreatic cancer (PANC-1) mice models. Tumor accumulation was quantified by gamma scintigraphy. Finally, an in vivo therapeutic efficacy study was performed in the CT26 tumor model, at suboptimal Gem dose < 20-40 mg/Kg [42].
Materials and Methods
Materials and Reagents
Hyaluronic acid (MW 51 kDa) was purchased from Lifecore Biomedical (USA). The Gemcitabine free base was purchased from LC Laboratories (USA). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), triethylamine (TEA), dipyridamole, anhydrous Dimethyl sulfoxide (DMSO), 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide (MTT) were purchased from Sigma (UK).4'-(Aminomethyl) fluorescein, hydrochloride (4'-AMF) was purchased from Life Technologies Limited, UK. Tris(hydroxypyridinone) amine (THP-amine) was synthesized in our labs as previously reported [43]. Snake Skin dialysis tubing (MWCO 10 kDa) was purchased from Thermo-fisher (USA). Anti-CD44 antibody (ab157107) was purchased from Abcam, UK. Dulbecco's Modified Eagle Medium (DMEM), Advanced RPMI-1640 medium, fetal bovine serum (FBS), penicillin/streptomycin, trypsin/EDTA, and phosphate buffered saline (PBS) were obtained from Gibco (Thermo Fisher Scientific Inc, UK). The radioactive probe [67Ga] was purchased from Mallinckrodt Pharmaceuticals (Netherlands) as 'Gallium Ga67 Citrate solution for injection' and the solution was used directly without further purification. Instant thin layer chromatography paper impregnated with silica gel (iTLC-SG) was obtained from Agilent Technologies (UK). BD flow cytometry tubes were obtained from VWR (UK). Isoflurane (IsoFlo) for anesthesia was purchased from Abbott Laboratories Ltd (UK). Heparinised micro-capillaries (5 µL, Hirschmann Laborgerate) were purchased from VWR (UK). Acetonitrile (ACN) for HPLC was purchased from S D Fine-Chem Limited India. All reagents were used without further purification.
Preparation of the Conjugates
HA sodium salt (51 kDa, 1 g) was dissolved in deionized water (20 mL), and dialyzed against a dilute HCl solution (pH 3.5) and then deionized water using a dialysis tube (MWCO 10 kDa), for 12 h each, followed by lyophilization. To prepare the HA-Gem conjugate, the desalted HA (600 mg, 1.5 mmol of carboxyl group) was dissolved in 6 mL of anhydrous DMSO under nitrogen. Subsequently, EDC (575 mg, 3 mmol) and NHS (345 mg, 3 mmol) dissolved in anhydrous DMSO (2 mL), were slowly added to the HA solution. The mixture was stirred for 1 h at room temperature to activate the HA carboxylic group. After 1 h, Gem (790 mg, 3 mmol) and TEA (418 μL, 3 mmol) were added to the activated HA solution. Subsequently, the solution was allowed to react for 24 h at room temperature. After the completion of the reaction, the reactant was purified by successive dialysis (MWCO 10 kDa) against an excess of 0.1 M NaCl, 25 % (v/v) ethanol solution and then deionized water. The solution was then lyophilized and stored at -20 °C until further use. A similar procedure was applied for the synthesis of HA-THP and HA-4'-AMF except that THP-amine and 4'-AMF were added in place of Gem. Details relating to these two reactions are provided in the supplementary information.
Characterization of the Conjugates
The synthesized conjugates were characterized by UV-visible spectrometry, nuclear magnetic resonance (1H NMR) and gel permeation chromatography (GPC). The Gem content in the HA-Gem conjugate was confirmed by UV (Perkin Elmer, Lambda 35, UV/VIS Spectrometer, USA) and the quantitative measurement was performed using a Gem calibration curve by measuring absorbance at 268 nm. For the analysis, the samples were dissolved in PBS (pH 7.4) and scanned in the range from 350 to 230 nm. The NMR spectra were recorded using deuterium oxide (D2O) as asolvent (Bruker 400 MHz, Bruker, Billerica, MA, USA). GPC analysis was performed using a Waters Chromatograph embedded with an ERC-7515A refractive index detector and Styragel® columns (7.8 mm x 300 mm x µμm). The detection range varied from 1 kDa to 10 MDa. The analysis was carried out using THF as an eluent at a flow rate of 1 mL/min. The THP-amine content in HA-THP conjugate was confirmed and quantified by UV-visible spectrometry. HA-4'-AMF was characterized by fluorescence spectroscopy as described in the supplementary information.
Stability of HA-Gem in Mouse Plasma
The stability of HA-Gem conjugate in mouse plasma was performed using a previously reported method with slight modification [29]. Gem (0.8 mg) or HA-Gem conjugate (equivalent to 0.8 mg of Gem) was added to 1 mL of plasma and incubated at 37 °C for 48 h. At scheduled time intervals, 100 μL of the plasma was taken out and replaced with the same amount of fresh plasma. Subsequently, 500 μL of ACN was added to 100 μL of each of the test plasma sample to precipitate the protein. The resulting mixture was vortexed for 5 min to extract the drug and centrifuged at 15000 rpm for 5 min at 4 °C. The supernatant was analyzed with RP-HPLC (SHIMADZU prominence, Japan), using a C-18 column (C-18G 250 x 4.5 mm), operated at 30°C with PDA detector (SPD-M20A) at a wavelength of 266 nm. The mobile phase consisted of ACN/water (70:30, v/v) with a flow rate of 1 mL/min. Drug concentrations were calculated from a Gem calibration curve prepared under the same conditions (Figure S1).
Hemolysis Assay
The hemolysis assay was performed according to our previously optimized method [44]. Fresh red blood cells (RBCs) were collected by centrifugation (3000 rpm, 10 min) from healthy human blood. The supernatant was discarded and the settled RBCs were washed three times with PBS (pH 7.4). The pellet containing RBC was further diluted with the PBS (1:10). The diluted RBC suspension (0.1 mL) was added to Gem or Gem-HA (19 and 38 μM) solutions, or HA at an equivalent concentration, prepared in the PBS (0.9 mL). Distilled water and PBS were used as positive (100 % hemolysis) and negative (0 % hemolysis) controls, respectively. Samples were incubated at 37 °C for 2h in a shaker-incubator. The samples were then centrifuged (4000 rpm, 5 min) to settle down the non-lysed RBCs. The supernatant was analyzed by using a microplate reader at 576 nm and the percent hemolysis was estimated by the formula:
% Hemolysis = 100(Abs-Abs0)/(Abs100-Abs0)
Where, Abs, Abs100, and Abs0 are the absorbance of the samples, negative control (100 % hemolysis) and positive control (0% hemolysis), respectively.
Cell Culture
The human pancreatic carcinoma PANC-1 (PANC-1; ATCC® CRL-1469TM) and PANC-0403 (PANC-0403; ATCC® CRL-2555TM) cells, CT26 murine colon carcinoma (CT26; ATCC, CRL-2638), and B16-F10 murine melanoma (B16-F10; ATCC, CRL-6475) were cultured in Advanced RPMI (1640, 1X) media. Human melanoma MDA-MB 435 cells (MDA-MB 435; ATCC® HTB-129) were cultured in DMEM. The media was supplemented with 10 % FBS, 1 % L-glutamine, 50 U/mL penicillin, 50 µg/mL streptomycin. The cells were routinely grown in 75 cm2 canted-neck tissue culture flask in an incubator at 37 °C in 5 % CO2 and passaged regularly by using Trypsin/EDTA when confluence of 90 % was reached and sub-cultured.
CD44 Expression Study
To evaluate the CD44 expression on the cell surface, PANC-1, PANC-0403, CT26, B16-F10 and MDA-MB 435 cells were collected by trypsinization and transferred into BD flow cytometer tubes. Cells (5 x 105 cells of each cell line) were incubated with rat anti-CD44 monoclonal antibodies (#ab119863, Abcam, UK) for 30 min at RT, washed twice with PBS and stained with goat anti-rat antibodies conjugated with PE (#ab7010, Abcam, UK) for 30 min at RT. Cells were then washed and fixed with 4 % PFA. At least 10,000 cells were gated and the fluorescence was measured using the 488 nm excitation line and the FL2 detector, BD FACS Calibur flow cytometer (BD Bioscience, USA). The extent of CD44 expression was expressed as mean fluorescence intensity (MFI).
In Vitro Cytotoxicity Studies
The cancerous cell lines PANC-1, PANC-0403, CT26, B16-F10 and MDA-MB 435 were seeded in 96-well plates and incubated with different concentrations of Gem or HA-Gem (200 μL of 0.01-100 μM) in complete RPMI media for 24 and 72 h. The cytotoxicity was examined using a MTT assay. In brief, at the end of the incubation period, media was removed from the wells and replaced with 120 µL of MTT solution at a final concentration of 0.5 mg/mL. After addition of MTT, cells were incubated for 3 h at 37 °C under 5 % CO2. At the end of the incubation, the supernatant was removed from the wells and formazan was dissolved in 200 µL of DMSO. The plate was read at 570 nm in an FLUO star OPTIMA plate reader (BMG Labtech) and the results were expressed as the percentage cell viability (mean ± SD and calculated using the following equation: % Cell viability = (A570 nm of treated cells/A570 nm of untreated control cells) × 100. A similar procedure was applied to evaluate the cytotoxicity of different concentrations of native HA (200 μL of 0.1-1000 μM). Percentage viability was calculated as a percentage of untreated cells and expressed as mean ± SD (n = 5). The statics were applied using two-way ANOVA following a Bonferroni post comparison test. A p value of <0.05 was considered statistically significant in all the studies.
Nucleoside Transporter Inhibition Assay
For the nucleoside transporter inhibition assay, CT26 and PANC-1 cells were seeded in 96- well plates. Dipyridamole was used as a nucleoside transporter inhibitor and the cells were pre-incubated with 10 μM dipyridamole for 30 min. Different concentrations of Gem or HA-Gem (0.01-100 μM) were added to the wells and the incubation was extended along with the inhibitor for 24 and 72 h, at 37 °C and 5 % CO2. At the end of the incubation period, media was removed from the wells and replaced with 120 µL of MTT solution. The MTT assay (as described above) was used to measure cell viability.
In Vitro Cellular Uptake
For cellular uptake, CT26 cells were seeded in advanced RPMI media overnight in 12-well plates at a density of 1 x 105 cells/well. The cells were incubated with 20 or 40 µg/mL of HA-4'-AMF for 1 or 4 h, at 37 °C and 5 % CO2. After incubation, cells were washed three times with PBS, trypsinized and washed with RPMI. The cells were centrifuged at 1500 rpm for 5 min and the pellet was re-suspended in 300 µL of PBS. The HA-4'-AMF fluorescence was measured using flow cytometry in which 10,000 cells were gated and fluorescence was measured in triplicate using the 488 nm excitation line and the FL1 and FL2 detector, BD FACS Calibur flow cytometer (BD Bioscience, USA).
Endocytosis Inhibition
To investigate the pathways by which HA-4´-AMF is taken up by cells, various uptake inhibitors were tested on PANC-1 and CT26 cells. Cells were seeded in 6-well plates at a density of 2 x 105 cells/well overnight. The cells were pre-incubated with either sodium azide (10 mM) and deoxyglucose (5 mM), as an energy-dependent metabolic inhibitor, or cytochalasin D (2 μM), as a macropinocytosis inhibitor, for 15 min. After that, 40 µg/mL of HA-4´-AMF was added to the wells and co-incubated for 1 h, at 37 °C and 5 % CO2. Subsequently, the cells were washed three times with PBS, trypsinized and collected by centrifugation at 1500 rpm for 5 min. The pellets were re-suspended in 300 μL PBS into BD flow cytometer tubes. The internalization of HA-4´-AMF was studied on 10,000 gated cells by measuring 4´-AMF fluorescence using a FL1 channel detector and a BD FACS Calibur flow cytometer (BD Biosciences). The measurements were performed in triplicate and presented as means ± SD (n=3).
CD44 Inhibition
For CD44 inhibition, PANC-1 and CT26 cells were seeded in 12 well plates at a density of 1 x 105 cells/well overnight. On the next day, the plates were kept on an ice bath for 10 min and 50 μg/mL of anti-CD44 (prepared in cold RPMI media) was added and left on ice 10 min. The plates were removed from the ice bath and 40 µg/mL of HA-4´-AMF was added to the wells and incubated for 1 h at 37 °C and 5 % CO2. After incubation, media was removed, washed three times with PBS, trypsinized and collected into BD flow cytometer tubes. The internalization of HA-4´-AMF in the cells was measured by flow cytometry as described previously.
Cell Cycle Analysis by Flow Cytometry
To examine cell cycle alterations induced by Gem and HA-Gem, flow cytometry analysis was performed. Briefly, CT26 cells were seeded in 12 well plates at a density of 1 × 105 cells/well overnight. The cells were treated with Gem (2 μM), HA-Gem (equivalent to 2 μM of Gem) or HA (equivalent amount) for 24 and 48 h. After treatment, cells were washed two times with PBS, trypsinized and collected into BD flow cytometer tubes. The cells were centrifuged 1500 rpm for 5 min, washed with PBS, re-suspended in 100 µL of PBS and 900 µL of 70% cold ethanol and placed at 4 °C for 1 h to fix and permeabilize the cells. Cells were then washed with PBS and treated with 50 µL of RNase (100 µg/mL in PBS). The DNA was stained with 400 µL of propidium iodide (PI) solution (40 µg/ml of PI in PBS) in the dark at 37 °C for 30 min. PI fluorescence was analyzed by flow cytometry using a BD FACS Calibur (BD Bioscience, US). Finally, 10,000 cells were gated and fluorescence was measured in triplicate for each condition by using the 488 nm excitation line and the FL2 detector and analyzed by FlowJo software. Results were expressed as averages of percentage cell populations in each phase of the cell cycle ± SD (n= 3).
Radiolabelling of HA-THP and Labelling Stability Studies
Radiolabelling of HA-THP with [67Ga] was performed by mixing equal volumes of HA-THP solution (1 mg/mL in H2O) and [67Ga] citrate salt solution (pH 7), at room temperature for 5 min. [67Ga] citrate alone, HA (1 mg/mL) and THP-amine (1 mg/mL) solutions underwent the same labelling conditions and were used as controls. All solutions were spotted onto TLC strips, developed in 0.1 M citrate (pH 4.5) as the mobile phase and allowed to dry before analysis. The auto-radioactivity of the strips was developed and counted quantitatively using a Cyclone phosphor detector (Packard Biosciences, PerkinElmer Inc., UK). The [67Ga]HA-THP solution was buffered with NaCl to achieve final NaCl concentration as 0.9 % prior to injection for in vivo studies. The stability of radiolabelling was examined by incubating the [67Ga]HA-THP citrate saline solution alone or mixing with an equal volume of FBS at 37 °C for 24 h, followed by TLC analysis.
Tumor Implantation for In Vivo Studies
All in vivo experiments were conducted under the authority of project and personal license granted by the UK Home Office and the UKCCCR Guidelines (1998). Female Balb/c mice aged 4-6 weeks (Envigo, UK) were inoculated subcutaneously with CT26 cells (1 × 106 cells in 0.1 mL PBS) in the lower flanks. Female NSG mice aged 4-6 weeks (Charles River, UK) were inoculated subcutaneously with PANC-1 cells (5 × 106 cells in 0.1 mL PBS) in the lower flanks. Nuclear imaging studies were conducted in mice carrying one tumor in one flank; biodistribution and therapy studies were performed in mice carrying two tumors one in each flank. Normal C57/Bl6 mice aged 4-6 weeks (Envigo, UK) without tumor inoculation were used for the assessment of the biodistribution of THP-amine.
Whole Body 3D SPECT/CT Imaging of [67Ga]HA-THP in CT26 Tumor-Bearing Mice Following Intravenous Injection
In vivo uptake of [67Ga]HA-THP following i.v. injection was examined firstly by 3D whole body SPECT/CT imaging. Two weeks after tumor inoculation, CT26 tumor-bearing mice were anesthetized by isoflurane inhalation and i.v. injected with 200 μg of [67Ga]HA-THP (1 mg/mL in citrate saline, 10 MBq). SPECT/CT imaging was carried out immediately, and at 4 h and 24 h after injection, on the same animal in the prone position using the Nano-SPECT/CT scanner (Bioscan, USA). The SPECT scans were acquired over 24 projections (60 s per projection), using a 4-head scanner with 1.4 mm pinhole collimators, for a total acquisition time of 30-40 min. CT scans were performed using a 45 kVP X-ray source after each SPECT imaging. SPECT images were reconstructed using HiSPECT (Scivis GmbH, Germany) and CT images were reconstructed using InVivoScopeTM, proprietary Bioscan software. Both images were fused and analyzed by InVivoScopeTM. SPECT/CT imaging was also carried out in CT26 tumor-bearing mice i.v. injected with [67Ga] or [67Ga]THP-amine (10 MBq in citrate saline) as controls to compare the biodistribution patterns.
Blood, Excretion and Organ Biodistribution Profiles of [67Ga]HA-THP in Two Tumor-Bearing Mice Models
Pharmacokinetic studies including blood circulation, excretion profiles and distribution in major organs of [67Ga]HA-THP were carried out in both CT26 tumor-bearing Balb/c mice and PANC-1 tumor-bearing NSG mice. When tumors reached the desired size (~ 200 mm2, roughly at two weeks and 5 weeks post-inoculation for CT26 tumors and PANC-1 tumors, respectively), mice were injected with [67Ga]HA-THP (200 µg HA-THP, ~1 MBq) via a tail vein. Blood samples were collected in heparinized micro-capillaries from 2 min up to 24 h post-injection. To obtain the excretion profiles, mice were housed singly in metabolic cages in which animals had free access to water but not food. After 24 h, urine and feces were collected from individual cages. To assess the biodistribution profiles, organs of principle interest including skin, liver, spleen, heart, lung, muscle, bone, brain, stomach, intestine and tumor were excised and weighed post mortem at 1, 4 and 24 h post-injection. The radioactivity of above tissues was counted by γ-scintigraphy (LKB Wallac 1282 Compugamma, PerkinElmer, UK). Results were calculated as percentage injected dose per organ (% ID/organ) or per gram organ (% ID/g) and expressed as mean ± SD (n=3). Normal C57/Bl6 mice aged 4-6 weeks without tumor inoculation were injected with [67Ga]THP-amine. The blood profile and organ biodistribution at 1 and 24 h were assessed.
In Vivo Anticancer Activity
The therapeutic efficacy of the HA-Gem conjugate was investigated using CT26 tumor-bearing Balb/c mice. Different concentrations of free Gem were tested in mice firstly to select the appropriate therapeutic dose for HA-Gem, at one week post tumor inoculation; mice were randomly divided into four groups: (i) untreated (control); (ii) 10 mg/kg Gem; (iii) 40 mg/kg Gem and (iv) 120 mg/kg Gem. Mice were anesthetized using isoflurane and injected every 4 days for a total of 4 doses. To determine the therapeutic action of Gem versus HA-Gem, at one week post tumor inoculation, mice were randomly divided into five groups (n = 10): (i) Untreated (control); (ii) 15 mg/kg Gem (free drug); (iii) 30 mg/kg Gem; (iv) 15 mg/kg HA-Gem and (v) 30 mg/kg HA-Gem. Mice were anesthetized using isoflurane and injected every 4 days via tail vein for a total of four doses. The tumor size and animal weight was measured three times each week. Animals were sacrificed when tumors reached 1000 mm3. Tumor sizes are presented as mean values ± SEM (standard error of the mean). Significant differences were examined using one-way ANOVA following a Bonferroni post comparison test for tumor growth delay studies. A p value of <0.05 was considered statistically significant in all the studies. Log-rank (Mantel-Cox) test was performed for survival analysis using Graph Pad Software.
Histological Examination of Major Organs
At the experimental end points of therapy studies, animals were sacrificed and major organs including heart, lung, liver, spleen and kidney were excised. The organs were rinsed with PBS and then immediately fixed in 10 % neutral buffered formalin as 5 mm2 pieces. Tissue sections were processed for Haematoxylin & Eosin (H&E) staining according to standard histological protocols at the Royal Veterinary College, UK. Stained sections were analyzed using a Leica DM 1000 LED Microscope (Leica Microsystems, UK) coupled with CCD digital camera (Qimaging, UK).
Statistical Analysis
The data were presented as mean ± S.D. for all experiments, except for therapy studies where standard error of the mean (S.E.M.) was used. n denotes the number of repeats. Statistical differences were examined using one-way ANOVA in which p < 0.05 was considered as significantly different in all studies. Statistical analysis for therapy, tumor growth and survival studies is described in the same section.
Results
Rationale of the Tripodal Design and Synthesis
In this study, we designed tripodal HA conjugates suitable for in vitro and in vivo therapy and imaging. The carboxylate group of HA was activated using EDC/NHS chemistry, prior to conjugation to the amino group of Gem (therapy), 4'-AMF (in vitro tracking) or THP-amine (for 67Ga chelation and in vivo imaging). The summary of the design is described in Scheme 1. Conjugates were purified by dialysis and the solid product obtained upon lyophilisation was stored at -20 °C until further use. The yield of HA-Gem, HA-4'-AMF and HA-THP conjugate was ~ 94 %, 91 %, and 90 %, respectively.
Scheme for synthesis of HA conjugates. The chemical synthesis scheme describing conjugation strategy of hyaluronic acid (HA) to Gemcitabine (Gem), 4'-(aminomethyl) fluorescein hydrochloride (4'-AMF), or tris(hydroxypyridinone) amine (THP-amine) using EDC/NHS chemistry.
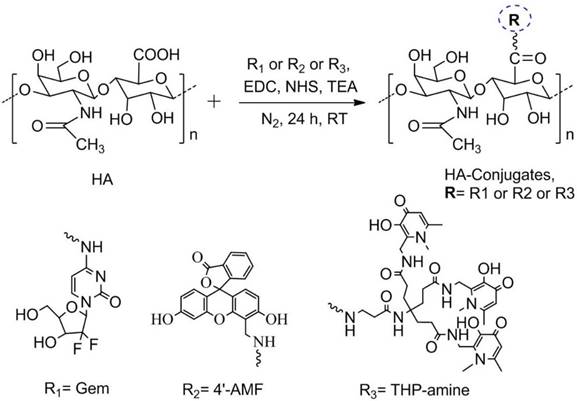
Confirmation of Gem Conjugation by 1H NMR and GPC
1H NMR spectroscopy was used to confirm Gem conjugation to HA (Figure 1). HA and HA-Gem present a peak at 1.88 ppm (-CH3-g of the acetamido moiety of N-acetyl-D-glucosamine). The characteristic Gem peaks are shown at 7.86 (-CH-b), 6.10 (-CH-a), 4.23 (-CH-d) and 3.94 (-CH-e). These peaks were also present in the dialyzed HA-Gem, confirming successful conjugation. The formation of HA-Gem conjugate was further confirmed by GPC analysis. The retention volumes of HA and HA-Gem were 19.49 mL and 15.28 mL, respectively (Figure 2). The reduction in HA retention volume upon conjugation was in line with previous studies [45, 46], supporting the NMR results, and confirming the successful conjugation of Gem to HA.
NMR spectra. 1H NMR (400 MHz) spectra of gemcitabine (Gem), hyaluronic acid (HA), and hyaluronic acid conjugated gemcitabine (HA-Gem) in D2O.
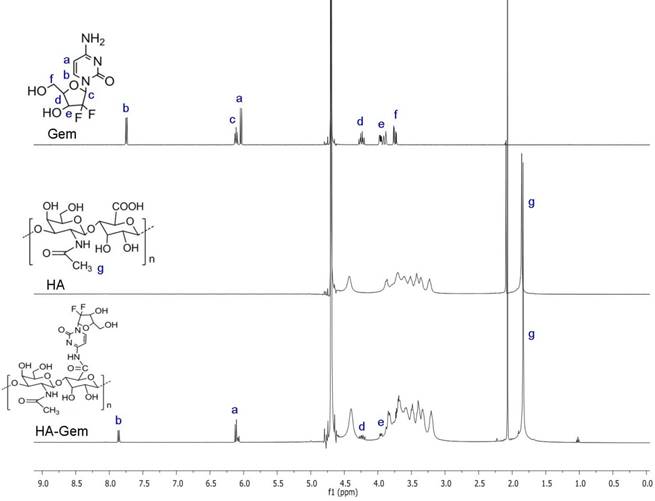
GPC analysis. GPC spectra of hyaluronic acid (HA) and HA conjugated Gemcitabine (HA-Gem). The analysis was performed with GPC equipped with ERC-7515A refractive index detector using a Styragel® column and THP as a mobile phase at elution rate of 1 mL/min. Retention times for HA and HA-Gem are 19.49 and 15.28 ml, respectively.
Quantification of Gem, 4'-AMF or THP in the Conjugates
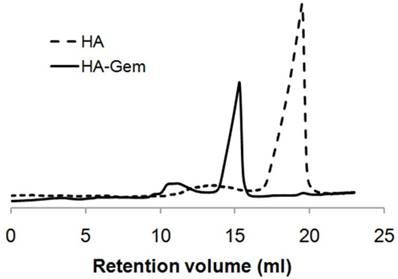
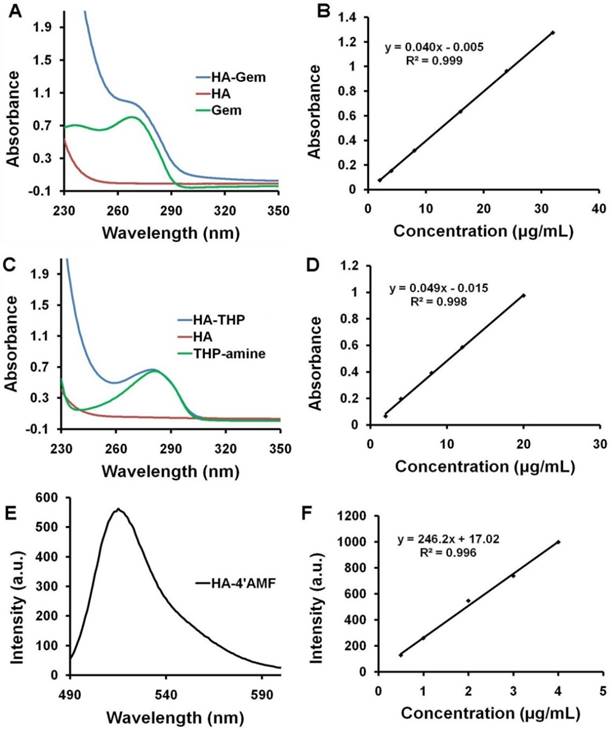
UV spectroscopy was used to quantify Gem, 4'-AMF or THP loading onto HA. Gem and THP-amine exhibit UV absorbance at 268 nm and 288 nm, respectively (Figure 3A, C). No peak shift was observed upon conjugation to HA. The fluorescence spectrum of 4'-AMF showed an emission maximum at 515 nm when excited at 480 nm (Figure 3E). Standard curves of Gem, 4'-AMF or THP (Figure 3B, D and F) were used to calculate the degree of substitution in HA-Gem, HA-4'-AMF or HA-THP which was calculated as ~38.15 µg/mg, 11.12 µg/mg or 10.45 µg/mg, respectively.
Absorption spectra and standard curves of the synthesized conjugates. (A) UV absorption spectra of HA, Gem and HA-Gem. (B) Standard curve of Gem in distilled water at 268 nm. (C) UV absorption spectra of HA conjugated tris(hydroxypyridinone) amine (HA-THP), HA and THP-amine. (D) Standard curve of THP-amine in PBS (pH 7.4) at 288 nm. (E) Fluorescence emission spectrum of HA conjugated 4'-(aminomethyl) fluorescein hydrochloride (HA-4'-AMF), at 480 nm excitation wavelength. (F) Standard curve of HA-4'-AMF (or 4'-AMF) in PBS (pH 10), at 480 nm and 515 nm excitation and emission wavelengths, respectively.
HA-Gem Exhibits Improved Stability and Sustained Drug Release in Mouse Plasma
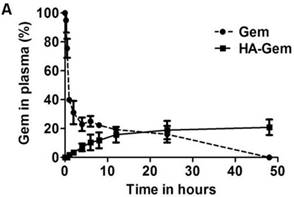
It is widely reported that native Gem is converted into its inactive metabolite 2′, 2′-difluorodeoxyuridine by cytidine deaminase [23], when administered via i.v. route. Contrary to Gem, which underwent enzymatic degradation following 37 °C incubation in plasma (23 % remaining at 4 h), HA-Gem showed sustained Gem release, with 7 % and 21 % drug being steadily released at 4 and 48 h of incubation time points (Figure 4A).
Ex vivo characterisation of the HA-Gem; in plasma for stability and in blood for hemolysis. (A) Time dependent estimation of Gem in plasma following release from HA-Gem. The free Gem or the conjugate was incubated with mouse plasma for 48 h at 37 °C. At scheduled time intervals, samples were taken and Gem quantified by RP-HPLC at a wavelength of 266 nm, mobile phase consisted of acetonitrile/water (70:30, v/v) and flow rate of 1 mL/min. (B) Ex vivo hemolytic activity studies on RBCs. The RBCs were incubated with Gem, HA-Gem, HA for 2 h at 37 °C. Distilled water and PBS (pH 7.4) were used as positive (100 % hemolysis) and negative controls (0% hemolysis), respectively. Data are reported as mean of three independent experiments ± SD, (***) P < 0.001, Gem versus HA-Gem.
HA-Gem Shows Lower Hemolytic Activity than Gem
Dose limiting hematopoietic toxicity was reported for Gem [47]. Gem conjugation to HA showed a significantly lower hemolytic activity at both concentrations tested (19 and 38 μM). Percentage hemolysis of 12 % and 33 %, 2 % and 6 % were reported for free Gem, HA-Gem at 19 and 38 μM, respectively (Figure 4B). As expected, no hemolytic activity was found for HA. The results suggest good in vitro hemocompatibility of HA-Gem, as previously suggested by others [48].
HA-Gem Is Active In Vitro in a Range of CD44-Expressing Cancer Cell Lines
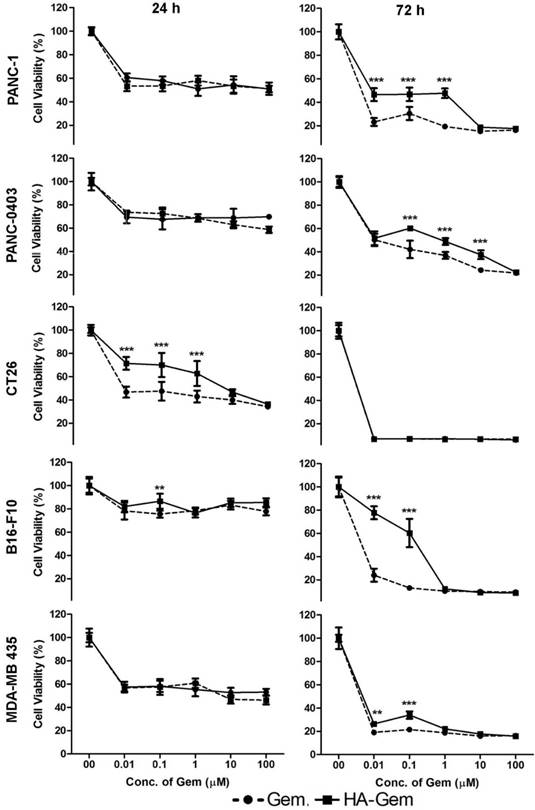
The cellular cytotoxicity of Gem and HA-Gem was evaluated by MTT assay in cancer cell lines expressing CD44 receptors to different degrees; PANC-1>CT26>PANC-0403> B16-F10 = MDA-MB 435 (Figure S2). Cells were incubated with Gem in the form of free drug or the conjugate, at 0.01-100 μM for 24 and 72 h. Dose- and time-dependent reductions in cell viability were observed in all cell lines (Figure 5). In all cell lines except CT26, no significant difference in cell viability between Gem and HA-Gem was observed at 24 h. Free Gem, however, showed slightly significantly higher cytotoxicity than HA-Gem at 72 h in most of the cell lines, presumably due to differences in internalization mechanisms and release kinetics compared to the free drug. CT26 showed the highest sensitivity amongst all the cell lines tested. No toxicity was observed for HA (without the drug) under conditions tested for Gem in PANC-1 and PANC-0403, the only cell lines so investigated (Figure S3).
In vitro cytotoxicity assay. Concentration dependent anticancer effect of Gemcitabine (Gem) and hyaluronic acid conjugated Gemcitabine (HA-Gem) was confirmed in a range of cancer cell lines (Pancreatic: PANC-1, PANC-0403; colon: CT26; melanoma: B16-F10; and breast: MDA-MB 435) using the MTT assay, at 24 h and 72 h post-incubation. Percentage viability was calculated as a percentage of untreated cells and expressed as mean ± SD (n = 5). * P < 0.05, ** P < 0.01, *** P < 0.001
Nucleoside Transporter Inhibition Reverses the Anticancer Activity of Gem, but not HA-Gem
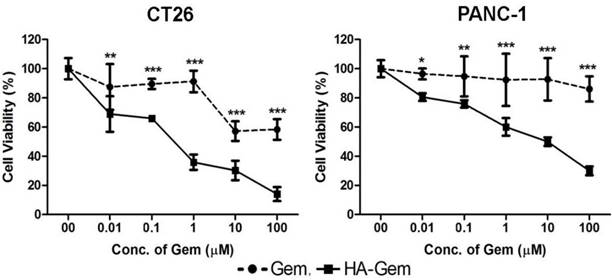
The activity of Gem is dependent on nucleoside transporters, which are required for internalization into cells. Reduced nucleoside transporter activity resulted in resistance to Gem therapy [49]. We have tested the effect of nucleoside transporter inhibition on the internalization of Gem, using 10 μM dipyridamole, which is a non-specific inhibitor of hENT1 transporters [50]. This resulted in significantly lower cytotoxicity for Gem but not the conjugate, in both cell lines tested; CT26 and PANC-1 (Figure 6 and Figure S4). This further confirmed that the free drug but not the conjugate is internalized into the cells using nucleoside transporters.
Nucleoside transporter inhibition assay. The effect of nucleoside transporter inhibition, using dipyridamol, on Gem and HA-Gem cytotoxicity was studied using a MTT assay, in CT26 and PANC-1 cells. Cells were pre-incubated with dipyridamol (10 µM) for 30 min, prior co-incubation with different concentration (0.01 to 100 μM) of Gem or HA-Gem for 72 h. Percentage viability was assessed by the MTT assay and calculated as a percentage of untreated cells and expressed as mean ± SD (n = 5). * P < 0.05, ** P < 0.01, *** P < 0.001 Gem versus HA-Gem.
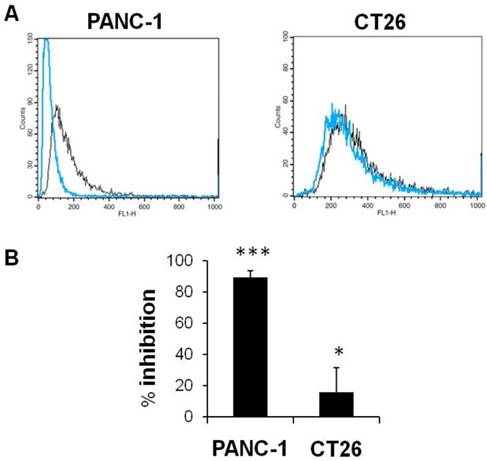
In Vitro Uptake of HA-4'-AMF in Colon CT26 and Pancreatic PANC-1 Cells is Macropinocytosis- and CD44-dependent
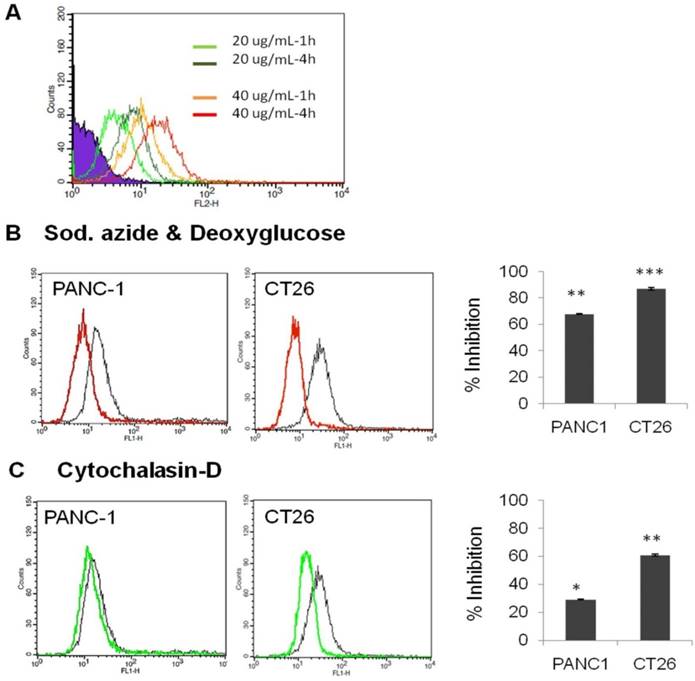
HA enters into cells via CD44 receptor-mediated endocytosis [36, 37, 39]. We tested the uptake of fluorescently labelled HA, HA-4'-AMF, in two CD44 expressing cell lines which exhibited the highest CD44 expression amongst the cell lines tested (PANC-1>CT26). The uptake of HA-4'-AMF in CT26 was dose- and time-dependent (Figure 7A). A general endocytosis inhibition condition, sodium azide (10 mM) and deoxyglucose (5 mM), resulted in inhibition of HA-4'-AMF uptake in both cell lines (Figure 7B). Similarly, Cytochalasin D (2 μM), a macropinocytosis inhibitor, resulted in uptake inhibition, more extensively in CT26 cells (Figure 7C). To determine if the uptake is CD44-receptor dependent, receptors were blocked with anti-CD44 (50 μg/mL) prior HA-4'-AMF treatment. This resulted in uptake inhibition, more especially in PANC-1 cells (Figure 8), in contrast to the Cytochalasin D effect. These studies concluded that both macropinocytosis and CD44 receptors are involved in HA uptake. CT26 appears to be more dependent on macropinocytosis than the CD44 receptor, for HA internalization, and vice versa for PANC-1.
Effect of endocytosis inhibitors on cellular uptake of HA-4´-AMF in vitro. (A) CT26 cells were incubated with the fluorescently labelled HA (HA-4´-AMF) for 1 or 4 h at 20 µg/mL or 40 µg/mL at 37 °C. Flow cytometry histograms confirmed that the intracellular uptake of HA-4'-AMF in CT26 cancer cells is time- and concentration-dependent. (B) The influence of general energy inhibition (sodium azide, 10 mM and deoxyglucose, 5 mM)) (red line). (C) Macropinocytosis (cytochalasin-D, 2 μM) (green line) on HA-4´-AMF uptake (black line). All treatments resulted in uptake inhibition. Cells were pre-incubated with either inhibitor for 15 min prior co-incubation with both the inhibitor and HA-4´-AMF (40 µg/mL for 1 h).
Effect of CD44 inhibition on cellular uptake of HA-4´-AMF in vitro. CT26 or PANC-1 cells were co-incubated with the fluorescently labelled HA (HA-4´-AMF) (40 µg/mL, black line) and anti-CD44 (50 μg/mL, blue line) for 1 h at 37 °C, to inhibit uptake via CD44 receptor. The cells were pre-treated with anti-CD44 for 10 min at 4 °C. (A) Flow cytometry histograms and (B) % inhibition indicate significant inhibition of the uptake in both cell lines.
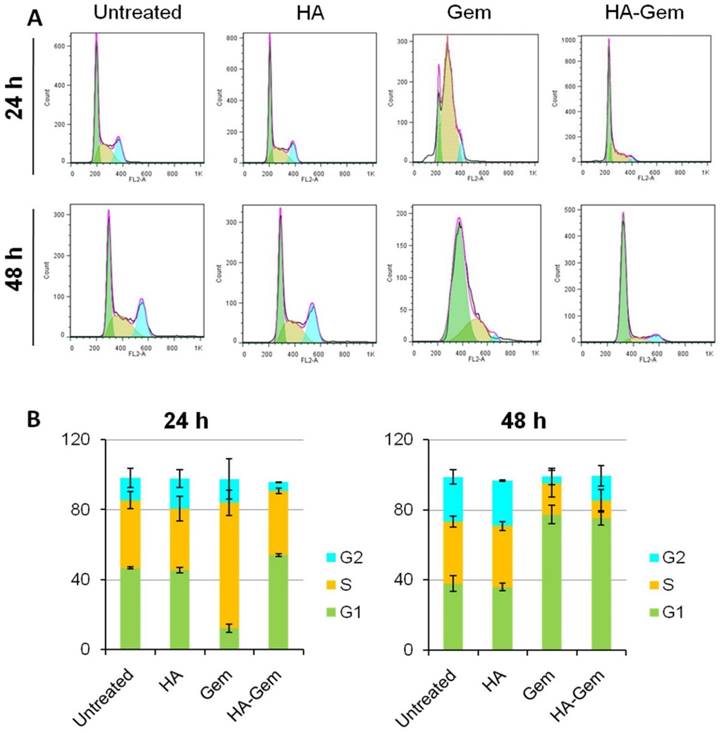
HA-Gem and Gem Shift the Cell cycle of CT26 from G1 to S phase
Cell cycle analysis was investigated at 24 and 48 h post-treatment (2 µM Gem concentration) using flow cytometry. Gem-treated cells showed ≈ 72 % of cells arrested in S phase after 24 h (Figure 9 A, B) while no apparent changes could be seen for Gem-HA or HA treatments, agreeing with the reduction of in vitro toxicity observed for Gem-HA with MTT the assay. However, cells treated for 48 h with Gem and HA-Gem showed a steady increase of ≈ 80 % of cells arrested in G1 phase (Figure 9 A, B). HA did not show any effect on the cell cycle analysis.
Cell cycle distribution analysis at 24 and 48 h in CT26 cells in vitro. The cells were seeded in 12 well plates at a density of 105 cells/well and then treated with (2 μM Gem concentration) free Gem or HA-Gem or HA for 24 and 48 h. (A) Representative histograms showing the cell cycle distribution and (B) Relative changes in the percentage of the cell cycle phases following 24 and 48 h indicate a shift in cell cycle distribution from G1 to S phase, which is more apparent at 24 h of Gem post-treatment. Cell cycle arrest in G1 phase was induced after 48 h post-treatment of both Gem and HA-Gem. Values are expressed as mean ± SD (n = 3).
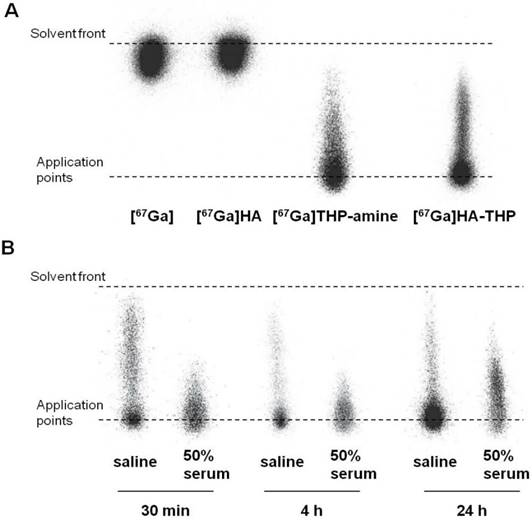
[67GA]-Radiolabelling of HA-THP Conjugate and Stability Studies
In this study, a rapid and efficient labelling reaction with the γ-emitter [67Ga] at neutral pH and room temperature was observed. As shown in the iTLC images (Figure 10A), free [67Ga] and [67Ga] HA alone appeared at the solvent front. Most of the radioactivity of [67Ga]THP-amine and [67Ga]HA-THP was observed at the application point. Some smeared through the ILC strips but did not travel to the solvent front, indicating the binding of [67Ga] with THP-amine, and the successful radiolabelling of HA-THP with [67Ga]. The labelling stability was examined by incubating equal volumes of [67Ga]HA-THP with saline or serum at 37 °C for 30 min, 4 h, and 24 h. iTLC results showed that the [67Ga]HA-THP remained stable without the observation of radioactivity spots at the solvent front (Figure 10B).
Radiolabelling of HA-THP-amine with [67Ga] and mouse serum stability studies. iTLC examination of (A) radiolabelling efficiency and (B) radiolabelling stability. Equal volume of HA-THP solution (1 mg/mL in H2O) and [67Ga] citrate solutions were mixed at room temperature for 5 min. [67Ga], HA (1 mg/mL), THP-amine (1 mg/mL) solutions were tested as controls. Labelling efficiency was analysed by iTLC using 0.1 M citrate (pH 4.5) as the mobile phase. For stability studies, [67Ga]HA-THP solution was diluted with saline to final NaCl concentration at 0.9 % (w/w). An aliquot of solution was mixed with an equal volume of FBS (50 % serum). Both solutions were kept at 37 °C and examined by iTLC after 30 min, 4 h and 24 h of incubation.
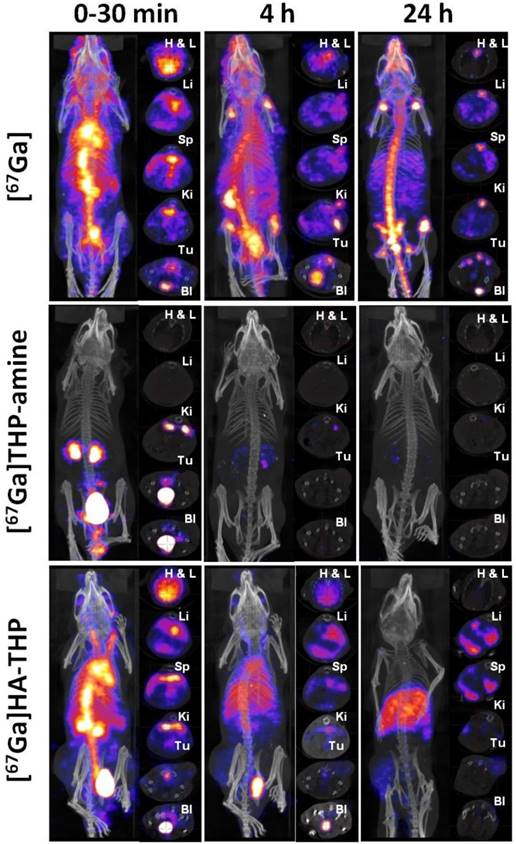
In Vivo Whole Body SPECT/CT Imaging
Whole body SPECT/CT images of uncomplexed [67Ga], [67Ga]THP-amine and [67Ga]HA-THP in CT26 tumor-bearing mice at multiple time points up to 24 h post-injection are shown in Figure 11. The three compounds showed marked differences in their in vivo biodistribution over the time course. [67Ga]THP-amine was rapidly cleared through urinary excretion since radioactivity was detected exclusively in kidney and bladder within 30 min after injection. This is in contrast to the distribution of [67Ga] where most of the activity was present in the circulation after injection, located throughout the whole mouse at 4 h, accumulated in the bone at 24 h. In the case of [67Ga]HA-THP, it displayed a relatively high concentration in the circulation but also exhibited renal excretion at early times post injection. The accumulation of [67Ga]HA-THP in liver, spleen and tumor was observed, and the uptake in CT26 tumor increased over time. No tumor uptake was detected for [67Ga] and [67Ga]THP-amine.
Whole body 3D SPECT/CT imaging of [67Ga], [67Ga]THP-amine and [67Ga]HA-THP in CT26 tumor-bearing Balb/c mice. Mice were i.v. injected with 10 MBq of [67Ga], [67Ga]THP-amine (200 µg) or [67Ga]HA-THP (200 µg). SPECT/CT imaging was carried out immediately after injection and at 4 h and 24 h post injection. The whole body image is presented in the left panel and the transverse images of selected organs are presented in the right panel. Abbreviations: Heart (H), lung (L), liver (Li), spleen (Sp), kidney (Ki), tumor (Tu) and bladder (Bl). In the [67Ga] treated group, tumor (Tu) and bladder (Bl) are captured in the same image, the bottom transverse images. Since the signals in spleen of [67Ga]THP-amine group were not detectable and spleen is not CT contrast organ, transverse images of spleen for these mice are not available.
In Vivo Tumor Uptake and Organ Biodistribution Studies of [67Ga]HA-THP in CT26 and PANC-1 Tumor-Bearing Mice
The organ biodistribution and tumor uptake profiles of [67Ga]HA-THP following i.v. injection were assessed quantitatively by γ-scintigraphy in two tumor models. As shown in Figure 12A-B, similar blood and excretion profiles were obtained in both tumor-bearing mice models. [67Ga]HA-THP showed long blood circulation time, in line with the SPECT/CT imaging results. More than 30 % of the injected dose (% ID) was detected in urine over 24 h in both models, whereas the amount detected in feces was invariably small. The uptake in major organs correlated well with SPECT/CT imaging results in which most [67Ga]HA-THP were detected in liver and spleen and the accumulation in these organs increased over time (Figure 12C-D, the left panel). The high radioactivity detected in the heart and lung at early time points (i.e. 1 and 4 h) was related to high blood concentrations. Uptake in kidney was monitored, suggesting urinary excretion of [67Ga]HA-THP. The tumor accumulation profiles of [67Ga]HA-THP between the two models were clearly different (Figure 12C-D, the right panel). Increasing uptake in CT26 tumors over time was observed in which the accumulation at 4 h and 24 h were significantly higher than 1 h post injection (p < 0.05 and p < 0.01, respectively) and the values were measured above 1 % ID/g of tumor. The uptake of [67Ga]HA-THP in PANC-1 tumor was lower, compared to CT26 tumors and the amounts (% ID/g of tumor) detected at different time points were not significantly different (< 1 % ID/g of tumor).
Blood, excretion and organ biodistribution profiles of [67Ga]HA-THP in CT26 tumor-bearing Balb/c mice and PANC-1 tumor-bearing NSG mice. (A) Blood concentrations and (B) excretion into urine and faeces, up to 24 h post injection. Major organ biodistribution at 1, 4 and 24 h post injection in (C) CT26 tumor-bearing mice and (D) PANC-1 tumor-bearing mice. Tumor uptake is shown to the right. Mice were i.v. injected with 1 MBq of [67Ga] HA-THP (200 µg). For excretion profiles, mice were housed individually in metabolic cages for 24 h immediately after injection. Results are expressed as % ID/organ or % ID/g of organ measured by γ-scintigraphy. Data is presented as mean ± S.D. (n=3). *p < 0.05; **p < 0.01.
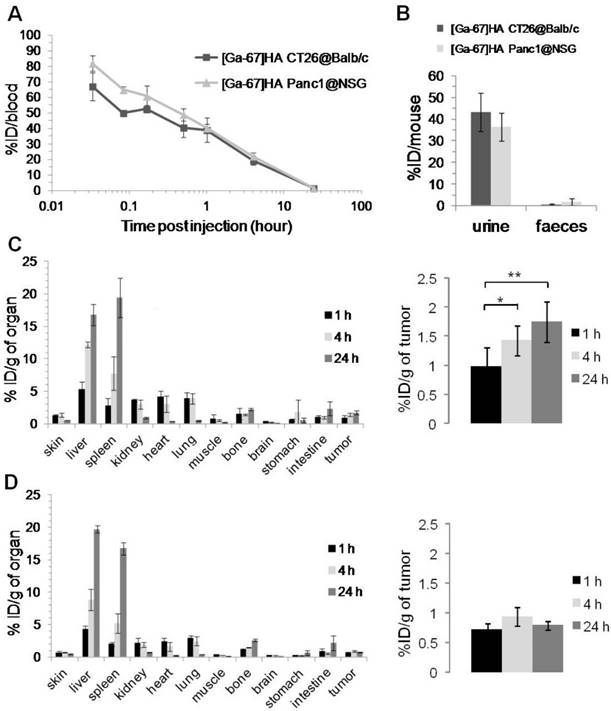
Blood and organ biodistribution profiles of [67Ga]THP-amine were also assessed in normal C57/Bl6 mice (Figure S5A-B). The results from quantitative γ-scintigraphy analyses correlated with the SPECT/CT imaging although using different mice strains. Fast clearance from the blood circulation and body was observed within the first-hour post injection. The organ biodistribution profiles of [67Ga]HA-THP in both models expressed as % ID/organ are expressed in Figure S5C and S5D respectively. The organ biodistribution at 1, 4 and 24 h post injection of HA in CT26 and PANC-1 bearing mice (µg (HA)/g tissue) is tabulated in Table S1.
Reduced HA-Gem Dose Delays Tumor Growth and Improves Survival of CT26 Tumor-Bearing Balb/c Mice
The anticancer potential of HA-Gem was investigated in CT26 tumor-bearing Balb/c mice. Firstly, the effective dose of Gem was determined in CT26 tumor-bearing mice following i.v. administration. Mice injected with 40 (21 ± 4 mm3) and 120 mg/kg (15 ± 3 mm3) of Gem exhibited significantly smaller tumor volume by day 20 (p < 0.05), when compared to 10 mg/kg Gem (291 ± 70 mm3) and untreated mice (453 ± 96 mm3) (Figure S6A). There were no significant differences in tumor volumes between untreated and 10 mg/kg Gem was observed. Changes in body weights remained within the 10 % weight loss range allowed in this type of treatment (Figure S6B). Suboptimal doses of 15 and 30 mg/kg of Gem were therefore selected for comparing the in vivo efficacy of Gem and its HA conjugate.
On day 28 post-therapy with Gem or HA-Gem, tumor volumes of mice treated with 15 mg/kg Gem or HA-Gem were 316 ± 83 or 61 ± 21 mm3 (p < 0.05), respectively, while 30 mg/kg treated resulted in 161 ± 68 and 25 ± 4 mm3 volumes for the respective treatments (p > 0.05) (Figure 13A). These results demonstrated that multiple injections of HA-Gem resulted in a significantly delayed tumor growth compared to the free drug at doses as low as 15 mg/kg). No significant changes in the body weights were observed following any of these treatments (Figure S7).
Evaluation of in vivo anti-tumor effect of Gem and HA-Gem in CT26 tumor bearing BALB/c mice. (A) In vivo anti tumor effect of Gemcitabine (Gem) and hyaluronic acid conjugated Gemcitabine (HA-Gem) in the CT26 tumor bearing BALB/c mice. (B) Percentage survival of mice. The mice were transplanted subcutaneously with 1 × 106 CT26 cells. The animals were then divided randomly into five groups: Untreated (control), 15 mg/kg Gem (free drug), 30 mg/kg Gem, 15 mg/kg HA-Gem and 30 mg/kg HA-Gem. Mice were intravenously administered a total of 4 injections on day 7, 10, 14 and 18 post-tumor inoculation (dotted vertical line) with the particular treatment. The size of the tumor was measured three times each week. Data are given as mean value ± SD (n = 8). One-way ANOVA was performed for tumor growth analysis following Bonferroni post comparison test. Log-rank (Mantel-Cox) test was performed for survival analysis using Graph Pad. *p < 0.05, **p < 0.01, ***p < 0.001.
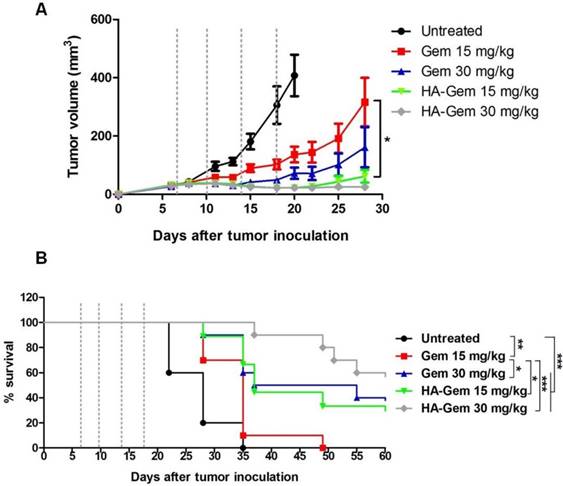
Log-rank survival analysis was carried out (Figure 13B). Interestingly, animals received HA-Gem showed significantly longer survival time compared to untreated mice or mice treated with free Gem.
No Histological Abnormalities are Found Post Therapy in Major Organs of CT26 Tumor-Bearing Mice
For histological examination, major organs including heart, lung, liver, spleen and kidney were excised at the experimental end points of therapy studies. The tissue sections were preceded for H&E staining. Despite the promising anti-tumor effect of HA-Gem, no obvious histological changes were observed in these major organs compared to untreated animals (Figure S8). The tissue sections from animals treated with free Gem showed no obvious histological abnormality either.
Discussion
Gem is one of the most effective anticancer drugs used for treatment several solid tumors [51]. However, rapid deamination leading to a short half-life necessitates its frequent administration in high doses to achieve chemotherapeutic efficacy [22, 23]. Moreover, Gem requires nucleoside transporters to aid its transport across cell membranes. This can be an issue in nucleoside transporter-deficient patients and can be associated with a high incidence of drug resistance. Preparation of macromolecular conjugates has been reported as one of the methods to tackle this problem [52].
Our study differs from previously reported studies on HA-based macromolecular delivery in a few aspects. In this study, we report a versatile HA conjugate amenable to modification to accommodate Gem (drug) or a nuclear imaging chelator. A HA-THP conjugate is synthesized for the first time. The THP chelator facilitates chelation of SPECT (67Ga) or PET (68Ga) imaging probes. The binding constant for [67Ga]-THP is high ensuring no leakage of [67Ga] from the complex. The high affinity also ensures a rapid and almost complete radiolabelling reaction. [67Ga]THP has been used to radiolabelling proteins [14]. Application of this radiolabelling strategy to a polymer such as HA is an important step towards designing HA-based conjugates for image-guided delivery.
HA pre-clinical imaging was reported previously with PET [53, 54] and CT/SPECT [55, 56] or MRI [57, 58] techniques. One study reported the distribution of [99mTc]-labelled HA into connective tissues after oral administration in rat [55]. In another study, tyramine-modified HA was radiolabelled with [125I] and its organ distribution examined following oral administration. Most of the conjugate remained localized in the stomach and intestine [56]. Two studies on PET imaging of HA-based nanocarriers, using either NOTA or DOTA chelators, were reported [53, 54]. In one PET imaging study, 1,4,7-triazacyclononane -1,4,7-triacetic acid (NOTA)-HA conjugate was synthesized and radiolabelled with 68Ga. PET imaging was carried out to detect myocardial infarction in rats. Higher accumulations of radiolabelled conjugate into the infarct than in healthy cardiac tissue was observed. This is due to the hyaluronan-receptor-induced targeting in the infarct area [53]. The closest to our study was PET imaging of HA-single-walled carbon nanotube hybrid, labelled 64Cu (HA-64Cu-DOTA-NTs) [54], which showed high tumor uptake in the CD44 positive, SCC7 tumor-bearing mouse model. This however is different from our study in that it is imaging the NTs functionalized with HA as opposed to HA as a macromolecular carrier. The organ biodistribution of HA polymer and HA-functionalized NTs is very different. The use of HA conjugated to THP, as a chelator, is totally novel. The combination of HA-Gem conjugate and HA-Gem conjugate offers qualitative tumor uptake data and potent treatment of CD44-expressing tumors in the same study.
To our knowledge, this is the first report which describes the preparation of HA-Gem conjugate that can be used for dual imaging and (mono) therapy of CD44-overexpressing tumors in vivo. It is expected that a mixture of HA-Gem and a trace of HA-THP can be administered to patients if image-guided delivery of this conjugate is to be implemented in clinical practice. Regulatory approval is likely to be more straightforward for 2 known chemical entities mixed in the same vial rather than the one conjugate containing both THP and Gem. The latter approach is likely to have more variability from one batch to another. Both Gem and THP were conjugated to HA via a relatively stable amide linkage, which is more stable than the ester linkage reported in the previous study [41]. Gem release from HA is expected to occur intra-cellularly, following CD44 mediated intra-cellular internalization. The molecular weight (MW) of HA chosen in this study (~51 kDa) has been reported to favor interactions with hyaluronan/CD44 receptor. Lower MW HA(< 30 kDa) exhibits lower affinity to the receptor [59] while higher MWHA forms a gel or precipitates from water [60]. The novelty of the performed study lies in many aspects including the method of synthesis, quantifying the targeting efficiency of HA in both highly vascularized murine colon cancer (CT26) and the highly resistant human pancreatic cancer (PANC-1) mice models and validating the use of HA-Gem conjugate as single agent for cancer therapy in vivo.
As expected, our study showed that Gem conjugation to HA protects it from degradation in plasma. This is possibly due to the involvement of the primary amine group in the amide bond formation, making it unavailable for the action of degradative enzymes. Following incubation of HA-Gem in plasma, a100 µL sample was withdrawn at 0, 15 min, 0.5, 1, 2 4, 6, 8, 12, 24 and 48 h, to which 500 µL of ACN was added to precipitate the protein and aid the extraction of the drug. An equal volume of fresh plasma was used to replace the release medium withdrawn. The results shown in Figure 4(A) demonstrated that the drug was slowly released from the conjugates in the presence of plasma. The slow Gem release from the conjugate, is beneficial as it is intended that Gem is not released prematurely in blood/plasma. After reaching to the target site, the drug can dissociate from the polymer due to the acidic microenvironment of the tumor [30]. Following internalization of HA-Gem into cells, the amide bond between polymer and drug can be enzymatically cleaved by the action of cathapsin B, a lysosomal protease. There are several reports on polymeric conjugates and prodrugs of gem that demonstrated the role of cathapsin B in cleavage of amide bond and release of drug inside the cells [30, 33, 61]. The conjugation results in reduced frequency of administration and reduced systemic side effects, i.e., hemolysis, and that a greater portion of HA-Gem is guaranteed to reach the tumor and target CD44 receptor. Thus, in principle, less frequent drug administration will be required. The slow release kinetics in serum was further supported by the lower cytotoxicity of HA-Gem than Gem observed in in vitro studies performed in the presence of mouse serum. It is expected that receptor-mediated uptake (HA-Gem) is a slower process than for free Gem (passive diffusion). Studies by other groups have shown reduced in vitro Gem cytotoxicity when conjugated to different polymers such as poly(ethyleneglycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate) [30] and poly-L-glutamic acid [62].
The nucleoside transporter inhibition study further confirmed that Gem and HA-Gem utilized two different internalization routes. This is in agreement with previous studies on Gem-phospholipid conjugates [63]. Interestingly our flow cytometry studies confirmed that HA internalization, as HA-4'-AMF, is energy dependent in both the cell lines tested: CT26 cells (colon) and PANC-1 cells (pancreatic). Cells, however, may favor one internalization route over another. Macropinocytosis was shown to play a more important role in HA internalization in CT26 than in PANC-1 cells. Cytochalasin D is a specific macropinocytosis inhibitor which stops the formation of actin filaments and membrane ruffling [64]. A previous study shared the similar findings in which HA endocytosed via macropinocytosis on B16-F10 melanoma cells [65]. Our study showed that blocking the receptor with an anti-CD44 antibody, reduced HA uptake in both cell lines, but more evidently in PANC-1 cells. Our results altogether confirmed HA internalization via CD44-mediated endocytosis, although this might not be the only mechanism involved [66-68].
The HA-THP conjugate was radiolabelled with γ-emitter [67Ga] in order to track its organ and tumor biodistribution, in the respective in vivo tumor models. THP-amine has been reported as an efficient bifunctional [68Ga] chelator for PET imaging [14, 43]. 67Ga has been in clinical use since 1980s for diagnostic imaging particularly for lymphoma. It has high affinity to blood and bone within hours after i.v. injection [69]. No free 67Ga was detected following incubation of [67Ga]THP-amine with human serum, at 37 °C for 4 h, followed by size exclusion chromatography [43]. The present study further demonstrated the outstanding binding affinity of 67Ga to both THP-amine and also HA-THP; as shown by the distinctly different SPECT imaging profiles of67Ga, [67Ga]THP-amine or [67Ga]HA-THP.
Gamma scintigraphy studies confirmed [67Ga]HA-THP targeting to CT26 and PANC-1 tumor following i.v. injection in mice (CT26>PANC-1). It is worth noting that there was a significant increase in tumor uptake over time, from 1 h to 24 h, in case the of CT26 tumors but PANC-1 showed steady values over time. This is not surprising as the murine CT26 tumor is a commonly used leaky and high vascular tumor model [70]. The human PANC-1 model on the other hand is poorly vascularized [71, 72]. The fact that PANC-1 cells express CD44 receptors at higher levels than CT26 suggests that tumor uptake in pancreatic cells can still occur, despite the compromised EPR effect in this tumor. Such difference in EPR may explain the increasing and steady tumor uptake pattern, over time, in CT26 and PANC-1 tumors, respectively. This indirect evidence of CD44-dependent uptake in PANC-1 cells in vivo complements the in vitro CD44-receptor inhibition data where PANC-1appeared to be more sensitive to uptake inhibition than CT26 cells.
Drug conjugation to HA is expected to improve its plasma profile and slow its clearance. Paclitaxel loaded HA-octadecyl micelles exhibited longer elimination half-life in the circulation, and higher AUC than Taxol® solution, in rats [73]. In another report, HA-quercetin conjugate showed a 20.1-fold and 4.9-fold increase in the half-life and AUC of quercetin, respectively, after i.v. administration in rat [74]. It has previously demonstrated that Gem modification on the N-4 position protected the drug from deactivation by degradative enzymes in plasma [29, 30], prolonged its circulation half-life [28, 29] and improved its therapeutic index [29-31]. To our knowledge, only one study reported the formulation of HA-Gem. In this study, Gem was conjugated to HA via an ester linkage. Two drug-polymer conjugates were co-administered: Gem-HA and paclitaxel- poly (L-lysine)-carboxylate [41]. A synergistic in vivo anticancer activity, in HuCCT1 biliary cancer, was obtained at a total of 6 i.v. injections, at twice weekly intervals and a dose of 108.80 µg (Gem) and 54 µg (paclitaxel) per mouse. In our study, the dose given was ~375 µg per mice which is higher than the dose reported in this published study [41]. It is however worth noting that a therapeutic response was achieved following a total of 4 injections, as a single agent, in our study, as opposed to 6 dose and co-administration of polymeric paclitaxel [41].
In our study, we have tested the cytotoxicity of free HA in CT26 and PANC-1cells in vitro, which was found to be non-cytotoxic up to 1000 µM. A number of in vivo reports suggested that HA has no effect on tumor growth rate. These studies were tested in SCC7 tumor bearing BALB/c nude mice [75], H22 bearing Kunming mice [74] and H22 bearing ICR mice [76].
To the best of our knowledge, this is the first time a HA-Gem conjugate has been reported as a single agent therapy. In our study, a 4-injection over a 28-day treatment period demonstrated that the HA-Gem delivered to CT26 tumors in vivo were sufficient to delay tumor growth and to prolong mice survival, at doses of 15 mg/kg (p< 0.05) and 30 mg/kg (p> 0.05) (Gem), compared to the free drug. A previous study, employed a liposomal Gem formulation in a CT26 tumor model, used a dose of 30-120 mg/kg for a total of 5 injections, repeated every third day [77]. No changes in body weight or histological abnormalities were seen confirming the biocompatibility of the conjugate. This result suggests that lowering the Gem dose, in the form of HA conjugate, can improve Gem therapeutic efficacy in vivo. This is likely to improve the therapeutic index of the drug as a whole and provide an effective treatment at a lower dose.
Conclusion
This work describes for the first time the synthesis of a tripodal HA conjugate, capable of SPECT imaging and cancer therapy in vivo. This is the first study reporting SPECT imaging of HA, as a macromolecular drug conjugate, in cancer applications. Furthermore, the use of THP, a Ga chelator, adds to the attractiveness of the approach as both SPECT (long-lived isotopes for pharmacokinetic studies) and PET (short-lived isotopes for clinical imaging studies) imaging can be performed. The fluorescently labelled conjugate provided information on the possible cell entry mechanisms employed in both CT26 and PANC-1 cells in vitro. Gamma scintigraphy and in vivo live imaging studies described the quantities of HA that reached the tumor, allowing for a correlation between tumor concentrations and therapeutic efficacy to be made. It was concluded that the HA-Gem conjugate was safe to use and resulted in improved therapeutic efficacy against CD44-expressing tumors, compared to the free drug, at significantly lower doses that what has been previously reported.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding from Department of Science and Technology, India and British council UK (Newton-Bhabha Fund) (DST/INSPIRE/NBHF/2014/14), Worldwide Cancer Research (12-1054) and EU FP7-ITN Marie-Curie Network programme RADDEL (290023) is acknowledged. K.-C.M. and N.H. are recipients of Graduate School International Research Award (GSIRA) and King's Health Partner's scholarships, respectively. R.D.D. is a Newton-Bhabha fellow.
Competing Interests
All the authors declare they have no competing of interests.
References
1. Delplace V, Couvreur P, Nicolas J. Recent trends in the design of anticancer polymer prodrug nanocarriers. Polymer Chemistry. 2014;5:1529-44
2. Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41:2545-61
3. Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev. 2013;42:1147-235
4. Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L. et al. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): a polymeric derivative of camptothecin (CPT). Br J Cancer. 2004;91:50-5
5. Ho DH, Brown NS, Yen A, Holmes R, Keating M, Abuchowski A. et al. Clinical pharmacology of polyethylene glycol-L-asparaginase. Drug Metab Dispos. 1986;14:349-52
6. Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R. et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83-94
7. Danhauser-Riedl S, Hausmann E, Schick HD, Bender R, Dietzfelbinger H, Rastetter J. et al. Phase I clinical and pharmacokinetic trial of dextran conjugated doxorubicin (AD-70, DOX-OXD). Invest New Drugs. 1993;11:187-95
8. Danson S, Ferry D, Alakhov V, Margison J, Kerr D, Jowle D. et al. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br J Cancer. 2004;90:2085-91
9. Rademaker-Lakhai JM, Terret C, Howell SB, Baud CM, De Boer RF, Pluim D. et al. A Phase I and pharmacological study of the platinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin Cancer Res. 2004;10:3386-95
10. Herzog T, Barret RJ, Edwards R, Oldham FB. Phase II study of paclitaxel poliglumex (PPX)/carboplatin (C) for 1st line induction and maintenance therapy of stage III/IV ovarian or primary peritoneal carcinoma. ASCO Annual Meeting Proceedings. 2005:5012
11. Therapeutics C. Improving Outcomes in PS2 Patients: results of the XYOTAXTM Phase III STELLAR trials. 11th World Congress on Lung Cancer, Barcelona, Spain, July. 2005
12. Yamamoto Y, Nishiyama Y, Monden T, Matsumura Y, Satoh K, Ohkawa M. Clinical usefulness of fusion of 131I SPECT and CT images in patients with differentiated thyroid carcinoma. Journal of Nuclear Medicine. 2003;44:1905-10
13. Bockisch A, Freudenberg LS, Schmidt D, Kuwert T. Hybrid imaging by SPECT/CT and PET/CT: proven outcomes in cancer imaging. Seminars in nuclear medicine: Elsevier. 2009:276-89
14. Ma MT, Cullinane C, Imberti C, Baguna Torres J, Terry SY, Roselt P. et al. New Tris(hydroxypyridinone) Bifunctional Chelators Containing Isothiocyanate Groups Provide a Versatile Platform for Rapid One-Step Labeling and PET Imaging with (68)Ga(3.). Bioconjugate chemistry. 2016;27:309-18
15. Ma MT, Cullinane C, Waldeck K, Roselt P, Hicks RJ, Blower PJ. Rapid kit-based 68Ga-labelling and PET imaging with THP-Tyr3-octreotate: a preliminary comparison with DOTA-Tyr3-octreotate. EJNMMI research. 2015;5:1-11
16. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7-12
17. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3-10
18. Garcia-Manteiga J, Molina-Arcas M, Casado FJ, Mazo A, Pastor-Anglada M. Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2',2'-difluorodeoxycytidine- induced cytotoxicity. Clin Cancer Res. 2003;9:5000-8
19. Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12:447-64
20. Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br J Cancer. 2007;97:145-51
21. Bender DM, Bao J, Dantzig AH, Diseroad WD, Law KL, Magnus NA. et al. Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J Med Chem. 2009;52:6958-61
22. Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB. et al. Cellular elimination of 2',2'-difluorodeoxycytidine 5'-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533-9
23. Reid JM, Qu W, Safgren SL, Ames MM, Krailo MD, Seibel NL. et al. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. Journal of clinical oncology. 2004;22:2445-51
24. Moog R, Burger AM, Brandl M, Schuler J, Schubert R, Unger C. et al. Change in pharmacokinetic and pharmacodynamic behavior of gemcitabine in human tumor xenografts upon entrapment in vesicular phospholipid gels. Cancer Chemother Pharmacol. 2002;49:356-66
25. Boven E, Schipper H, Erkelens CA, Hatty SA, Pinedo HM. The influence of the schedule and the dose of gemcitabine on the anti-tumour efficacy in experimental human cancer. British journal of cancer. 1993;68:52
26. Dasanu CA. Gemcitabine: vascular toxicity and prothrombotic potential. Expert opinion on drug safety. 2008;7:703-16
27. Hui YF, Reitz J. Gemcitabine: a cytidine analogue active against solid tumors. Am J Health Syst Pharm. 1997;54:162-70
28. Vandana M, Sahoo SK. Long circulation and cytotoxicity of PEGylated gemcitabine and its potential for the treatment of pancreatic cancer. Biomaterials. 2010;31:9340-56
29. Tao XM, Wang JC, Wang JB, Feng Q, Gao SY, Zhang LR. et al. Enhanced anticancer activity of gemcitabine coupling with conjugated linoleic acid against human breast cancer in vitro and in vivo. Eur J Pharm Biopharm. 2012;82:401-9
30. Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Self-assembling, amphiphilic polymer-gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjug Chem. 2013;24:1161-73
31. Maksimenko A, Mougin J, Mura S, Sliwinski E, Lepeltier E, Bourgaux C. et al. Polyisoprenoyl gemcitabine conjugates self-assemble as nanoparticles, useful for cancer therapy. Cancer Lett. 2013;334:346-53
32. Brusa P, Immordino ML, Rocco F, Cattel L. Antitumor activity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Anticancer Res. 2007;27:195-9
33. Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100:331-46
34. Chung WG, Sandoval MA, Sloat BR, Lansakara PD, Cui Z. Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1. J Control Release. 2012;157:132-40
35. Lee MS, Lee JE, Byun E, Kim NW, Lee K, Lee H. et al. Target-specific delivery of siRNA by stabilized calcium phosphate nanoparticles using dopa-hyaluronic acid conjugate. J Control Release. 2014;192:122-30
36. Dosio F, Arpicco S, Stella B, Fattal E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv Drug Deliv Rev. 2016;97:204-36
37. Galer CE, Sano D, Ghosh SC, Hah JH, Auzenne E, Hamir AN. et al. Hyaluronic acid-paclitaxel conjugate inhibits growth of human squamous cell carcinomas of the head and neck via a hyaluronic acid-mediated mechanism. Oral Oncol. 2011;47:1039-47
38. Mittapalli RK, Liu X, Adkins CE, Nounou MI, Bohn KA, Terrell TB. et al. Paclitaxel-hyaluronic nanoconjugates prolong overall survival in a preclinical brain metastases of breast cancer model. Mol Cancer Ther. 2013;12:2389-99
39. Goodarzi N, Ghahremani MH, Amini M, Atyabi F, Ostad SN, Shabani Ravari N. et al. CD44-targeted docetaxel conjugate for cancer cells and cancer stem-like cells: a novel hyaluronic acid-based drug delivery system. Chem Biol Drug Des. 2014;83:741-52
40. Banzato A, Bobisse S, Rondina M, Renier D, Bettella F, Esposito G. et al. A paclitaxel-hyaluronan bioconjugate targeting ovarian cancer affords a potent in vivo therapeutic activity. Clin Cancer Res. 2008;14:3598-606
41. Noh I, Kim HO, Choi J, Choi Y, Lee DK, Huh YM. et al. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763-74
42. Zheng SJ, Zheng SP, Huang FY, Jiao CL, Wu RL. Synergistic anti-tumor effect of recombinant chicken fibroblast growth factor receptor-1-mediated anti-angiogenesis and low-dose gemcitabine in a mouse colon adenocarcinoma model. World J Gastroenterol. 2007;13:2484-9
43. Berry DJ, Ma Y, Ballinger JR, Tavare R, Koers A, Sunassee K. et al. Efficient bifunctional gallium-68 chelators for positron emission tomography: tris(hydroxypyridinone) ligands. Chem Commun (Camb). 2011;47:7068-70
44. Dubey RD, Alam N, Saneja A, Khare V, Kumar A, Vaidh S. et al. Development and evaluation of folate functionalized albumin nanoparticles for targeted delivery of gemcitabine. Int J Pharm. 2014;492:80-91
45. Vandana M, Sahoo SK. Synergistic activity of combination therapy with PEGylated pemetrexed and gemcitabine for an effective cancer treatment. Eur J Pharm Biopharm. 2015;94:83-93
46. Vandana M, Sahoo SK. Reduced folate carrier independent internalization of PEGylated pemetrexed: a potential nanomedicinal approach for breast cancer therapy. Mol Pharm. 2012;9:2828-43
47. Rattmann I, Kleff V, Sorg UR, Bardenheuer W, Brueckner A, Hilger RA. et al. Gene transfer of cytidine deaminase protects myelopoiesis from cytidine analogs in an in vivo murine transplant model. Blood. 2006;108:2965-71
48. Gupta A, Asthana S, Konwar R, Chourasia MK. An insight into potential of nanoparticles-assisted chemotherapy of cancer using gemcitabine and its fatty acid prodrug:a comparative study. J Biomed Nanotechnol. 2013;9:915-25
49. Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA. et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349-57
50. Mey V, Giovannetti E, De Braud F, Nannizzi S, Curigliano G, Verweij F. et al. In vitro synergistic cytotoxicity of gemcitabine and pemetrexed and pharmacogenetic evaluation of response to gemcitabine in bladder cancer patients. Br J Cancer. 2006;95:289-97
51. Abdayem P, Ghosn M, Valero V, Walters R, Arun B, Murray JL. et al. Phase I and II Study of Gemcitabine and Vinorelbine in Heavily Pretreated Patients with Metastatic Breast Cancer and Review of the Literature. J Cancer. 2014;5:351-9
52. Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688-701
53. Jadhav S, Kakela M, Makila J, Kiugel M, Liljenback H, Virta J. et al. Synthesis and In Vivo PET Imaging of Hyaluronan Conjugates of Oligonucleotides. Bioconjug Chem. 2016;27:391-403
54. Swierczewska M, Choi KY, Mertz EL, Huang X, Zhang F, Zhu L. et al. A facile, one-step nanocarbon functionalization for biomedical applications. Nano Lett. 2012;12:3613-20
55. Balogh L, Polyak A, Mathe D, Kiraly R, Thuroczy J, Terez M. et al. Absorption, uptake and tissue affinity of high-molecular-weight hyaluronan after oral administration in rats and dogs. J Agric Food Chem. 2008;56:10582-93
56. Ma S-Y, Nam YR, Jeon J, Rho JK, Lee D-E, Choi DS. et al. Simple and efficient radiolabeling of hyaluronic acid and its in vivo evaluation via oral administration. Journal of Radioanalytical and Nuclear Chemistry. 2015;305:139-45
57. Wu G, Zhang H, Zhan Z, Lu Q, Cheng J, Xu J. et al. Hyaluronic Acid-Gadolinium Complex Nanospheres as Lymphatic System-Specific Contrast Agent for Magnetic Resonance Imaging. Chinese Journal of Chemistry. 2015;33:1153-8
58. Moon M, Thomas RG, Heo S-u, Park M-S, Bae WK, Heo SH. et al. A Hyaluronic Acid-Conjugated Gadolinium Hepatocyte-Specific T1 Contrast Agent for Liver Magnetic Resonance Imaging. Molecular Imaging and Biology. 2015;17:497-503
59. Wolny PM, Banerji S, Gounou C, Brisson AR, Day AJ, Jackson DG. et al. Analysis of CD44-hyaluronan interactions in an artificial membrane system: insights into the distinct binding properties of high and low molecular weight hyaluronan. J Biol Chem. 2010;285:30170-80
60. Fan X, Zhao X, Qu X, Fang J. pH sensitive polymeric complex of cisplatin with hyaluronic acid exhibits tumor-targeted delivery and improved in vivo antitumor effect. Int J Pharm. 2015;496:644-53
61. Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaele D. et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6:2544-8
62. Kiew LV, Cheong SK, Sidik K, Chung LY. Improved plasma stability and sustained release profile of gemcitabine via polypeptide conjugation. Int J Pharm. 2010;391:212-20
63. Alexander RL, Greene BT, Torti SV, Kucera GL. A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother Pharmacol. 2005;56:15-21
64. Huth US, Schubert R, Peschka-Suss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110:490-504
65. Greyner HJ, Wiraszka T, Zhang LS, Petroll WM, Mummert ME. Inducible macropinocytosis of hyaluronan in B16-F10 melanoma cells. Matrix Biol. 2010;29:503-10
66. Tammi R, Rilla K, Pienimaki J-P, MacCallum DK, Hogg M, Luukkonen M. et al. Hyaluronan enters keratinocytes by a novel endocytic route for catabolism. J Biol Chem. 2001;276:35111-22
67. Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C. et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282:16667-80
68. Ohno S, Im HJ, Knudson CB, Knudson W. Hyaluronan oligosaccharides induce matrix metalloproteinase 13 via transcriptional activation of NFkappaB and p38 MAP kinase in articular chondrocytes. J Biol Chem. 2006;281:17952-60
69. Sephton RG, Hodgson GS, De Abrew S, Harris AW. Ga-67 and Fe-59 distributions in mice. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 1978;19:930-5
70. Green CE, Liu T, Montel V, Hsiao G, Lester RD, Subramaniam S. et al. Chemoattractant signaling between tumor cells and macrophages regulates cancer cell migration, metastasis and neovascularization. PLoS One. 2009;4:e6713
71. Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M. et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104:3460-5
72. Sofuni A, Iijima H, Moriyasu F, Nakayama D, Shimizu M, Nakamura K. et al. Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J Gastroenterol. 2005;40:518-25
73. Liu Y, Sun J, Lian H, Li X, Cao W, Bai L. et al. Determination of paclitaxel in hyaluronic acid polymeric micelles in rat blood by protein precipitation-micelle breaking method: application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;935:10-5
74. Pang X, Lu Z, Du H, Yang X, Zhai G. Hyaluronic acid-quercetin conjugate micelles: synthesis, characterization, in vitro and in vivo evaluation. Colloids Surf B Biointerfaces. 2014;123:778-86
75. Lee JY, Chung SJ, Cho HJ, Kim DD. Iodinated hyaluronic acid oligomer-based nanoassemblies for tumor-targeted drug delivery and cancer imaging. Biomaterials. 2016;85:218-31
76. Yang C, Wang X, Yao X, Zhang Y, Wu W, Jiang X. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J Control Release. 2015;205:206-17
77. Lim SK, Shin DH, Choi MH, Kim JS. Enhanced antitumor efficacy of gemcitabine-loaded temperature-sensitive liposome by hyperthermia in tumor-bearing mice. Drug Dev Ind Pharm. 2014;40:470-6
Author contact
![]() Corresponding author: Professor Khuloud T Al-Jamal, 150 Stamford Street, Institute of Pharmaceutical Science, King's College London, Franklin-Wilkins Building, 150 Stamford Street, London SE1 9NH, UK. E-mail: khuloud.al-jamalac.uk.
Corresponding author: Professor Khuloud T Al-Jamal, 150 Stamford Street, Institute of Pharmaceutical Science, King's College London, Franklin-Wilkins Building, 150 Stamford Street, London SE1 9NH, UK. E-mail: khuloud.al-jamalac.uk.