- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Early infection mechanism of...
3. Virus-inspired nanocarriers
4. Bio-nanocapsule
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(4):415-429. doi:10.7150/ntno.21723 This issue Cite
Review
Current Progress of Virus-mimicking Nanocarriers for Drug Delivery
Masaharu Somiya*, Qiushi Liu*, Shun'ichi Kuroda ![]()
The Institute of Scientific and Industrial Research, Osaka University, Osaka 567-0047, Japan
*These authors contributed equally to this work.
Received 2017-7-2; Accepted 2017-10-9; Published 2017-10-31
Abstract

Nanomedicines often involve the use of nanocarriers as a delivery system for drugs or genes for maximizing the therapeutic effect and/or minimizing the adverse effect. From drug administration to therapeutic activity, nanocarriers must evade the host's immune system, specifically and efficiently target and enter the cell, and release their payload into the cell cytoplasm by endosomal escape. These processes constitute the early infection stage of viruses. Viruses are a powerful natural nanomaterial for the efficient delivery of genetic information by sophisticated mechanisms. Over the past two decades, many virus-inspired nanocarriers have been generated to permit successful drug and gene delivery. In this review, we summarize the early infection machineries of viruses, of which the part has so far been utilized for delivery systems. Furthermore, we describe basics and applications of the bio-nanocapsule, which is a hepatitis B virus-mimicking nanoparticle harboring nearly all activities involved in the early infection machineries (i.e., stealth activity, targeting activity, cell entry activity, endosomal escaping activity).
Keywords: biomimicking, bio-nanocapsule, endosomal escape, hepatitis B virus, membrane fusion, virus-inspired nanomedicine.
1. Introduction
The drug delivery system (DDS) has been studied as a means of improving medication. Since the “magic bullet” concept was described about a hundred years ago by Paul Ehrlich [1], scientists in medicine, pharmaceutical, and material science have been trying to discover or create sophisticated drugs that target specific molecules, cells, and tissues. DDS is one of the key technologies that can deliver drugs to target sites in the body. Numerous types of nanomaterials, including liposomes (LPs) and polymers, have been investigated as a DDS, some of which are now used in clinical settings [2-4]. These nanomaterials contain drug molecules by encapsulation, physical adsorption, or chemical linkage, and deliver them to the target. Nanoformulation of a drug can avoid renal excretion and allow prolonged circulation in the body [5], which contributes to improved bioavailability. Moreover, nano-sized materials can passively accumulate at tumor tissues or at sites of inflammation site via the enhanced permeability and retention (EPR) effect [6]. When the nanoformulation contains a targeting moiety, it can recognize specific target molecules in the body and accumulate at the specific site in the body [7].
However, some researchers have cautioned that the current DDS technology remains far from the true “magic bullet”. One meta-analysis revealed that only 0.7% of total nanoparticles (NPs) administered in vivo accumulates at the target tumor tissues [8]. Almost all (over 99%) of the drugs accumulate at non-target tissues or are cleared from body. Furthermore, in the clinical setting, human tumors sometimes do not exhibit an EPR effect [9]. Current DDS studies have been summed up as “many papers, few drugs” [10,11]. This situation reflects the complexity of nanocarriers compared to parental drugs. Furthermore, despite the contrary expectation, active targeting DDS only slightly improves the pharmacokinetics and accumulation of drugs into target tissues compared to non-targeting formulations. Cytoplasmic delivery is another issue of DDS. For the delivery of nucleic acid medicines, such as plasmid DNA, short interfering RNA (siRNA), and microRNA (miRNA), drug molecules should be delivered to the nucleus or cytoplasm of target cells to exert their biological function [12]. Nucleic acids do not inherently penetrate cell membranes and have to be intracellularly delivered via a DDS. After the cellular uptake of these drugs, endosomal escape is a rate-limiting step [13]. Even in the cutting-edge lipid NP platform, only 1 to 2% of cargo siRNA escapes from the endosome to the cytoplasm, to elicit RNA interference [14].
While synthetic nanocarriers remain hampered by these issues, viruses have naturally evolved to deliver genetic materials (DNA or RNA genome) into host cells in an elaborate and rational fashion. After the entry into host body, viruses can evade host defense and reach specific organs or cells. Once viruses are taken up by cells, they traffic the intracellular space and release their genome to replicate themselves. Viruses exploit their protein and lipid components to surmount cellular barriers. DDS technology can learn a great deal from viruses. Bio-inspired materials are being investigated for a wide variety of applications, such as in material science and biomedical research. These materials have unique characteristics and functions compared to human-designed materials [15]. Recombinant viruses have already been utilized as delivery systems in gene therapy [16]. However, natural virus-based DDS has inherent safety issues, including immunogenicity and insertion of genetic information of virus into host genome. Several clinical trials failed due to the safety problem of viral vectors [17]. Ideally, the infection machinery should be transplanted from viruses to artificial NPs to avoid the intrinsic and unexpected toxicity of viruses. In this review, we summarize the machinery that operates early in the viral infection process, which can hopefully be utilized as a virus-inspired DDS. We focus on the evasion of the reticuloendothelial system (RES), tissue tropism, cell entry, and endosomal escape. Current approaches to develop virus-inspired DDSs are described. In the final section of this review, we summarize our strategy to develop virus-inspired DDS by mimicking the infection machinery of hepatitis B virus (HBV).
2. Early infection mechanism of viruses
2.1. Immune system evasion
For the viruses, the first barrier of the host is the immune system. The immune system involves monocytes, macrophages, dendritic cell, and the complement system [18]. This is a complicated and rigorous system involving coordinate function of the aforementioned systems in the clearance of foreign substances. On the other hand, millions of years of evolution have equipped viruses with strategies to evade humoral and cellular immune responses [19,20]. Many of these stealth activities are determined by viral physiochemical characteristics that include size, shape, hydrophobicity, and surface charge. In addition to these physicochemical characteristics, viral proteins may function to circumvent the immune reaction of the host [21-23].
2.1.1. Physiochemical characteristics
Viruses have a variety of sizes and shapes, which can determine their ability to evade the host immune response. In general, viruses can be divided into two categories by shape. Spherical viruses include human immunodeficiency virus 1 (HIV-1), influenza virus (IFV), HBV, and adeno associated virus. Non-spherical viruses are rod-like or elliptical in shape (e.g., tobacco mosaic virus (TMV)). Most viruses are spherical, although different subviral structures can be present. For example, HBV specifically infects human liver cells. The virion is an approximately 42 nm-sized spherical structure termed as the Dane particle [24]. Subviral particles of HBV can be found in the patients' plasma. Interestingly, different from the Dane particle, another particle is spherical with an approximate diameter of 22 nm and still another particle is filamentous and lacks genetic materials. The biological function of both particles is unclear [25]. Recently, it was postulated that subviral particles may be a decoy for the neutralizing antibodies produced against the HBV virion [26].
Biodistribution, especially the circulation time in bloodstream, has been evaluated. Concerning plant virus, the TMV is rod-shaped, 300 nm in length, and 18 nm in diameter. At lower viral protein concentrations, TMV can form a 50-nm spherical structure. In mice, rod-shaped structures have a longer circulating time in bloodstream and are cleared less rapidly from tissues than spherical-shaped structures, indicating that viral shape plays an important role in biodistribution [27,28]. However, the effect of natural viruses' shape on their immune evasion has been difficult to study. To overcome this barrier, different methods have evaluated viral behavior in the bloodstream, including virus-mimicking or gold NPs as a model. The intent of this model is to simulate the features that could influence immune evasion [29,30]. For example, rod-shaped gold NPs modified with poly (ethylene glycol) (PEG) accumulate in the liver to a lesser extent, and circulate longer in the blood compared to spherical NPs [31].
Size of viral particle is also important for host evasion. Monocytes, macrophages, and other phagocytic cells that are responsible for the clearance of viruses [32], efficiently engulf particles in size-dependent manner, especially particles in the micrometer range [33]. Indeed, the diameter of virus influences the cellular uptake pathway. Large viruses such as mimivirus (~760 nm in diameter) are taken up by macrophages via phagocytosis [34], while smaller viruses tend to be internalized to cells via clathrin-mediated endocytosis [35]. Although immune evasion mechanism of each virus is different, size may be one of the key factors that determines immune evasion ability of viruses.
Surface charge and hydrophobicity of viral particles are also important for immune escape. Hydrophobic particles are more likely to be taken up by cells than hydrophilic, non-ionic particles [36]. Positively charged particles have longer circulatory times than negatively charged particles, indicating that surface charge influences the circulation time of viruses [37]. The surface charge of virions depends on the surrounding environment. According to the isoelectric point (pI) of viral protein, which is governed by the pH of the environment, surface charge of viral protein is variable, even from the negative charge to positive charge. Among the 104 viruses investigated, most of the viruses have less than 7 of pI, and viruses possessing pI higher than 8 are rarely found [38], suggesting that majority of viruses show negatively charged surfaces in physiological condition. Viruses with negatively charged glycans may prevent the opsonization, leading to the escape from phagocytosis [39].
2.1.2. Viral protein
Viruses use many complicated mechanisms to evade the host immune system in a viral protein-mediated manner [20,40]. The strategy of evasion mechanism includes both interference with antigen presentation by major histocompatibility complex (MHC) molecules and evasion from cytotoxic T lymphocytes (CTLs) by antigenic variation. The CTLs recognize viral antigens on the MHC class I and activate antiviral immune system [41]. Some viruses utilize these unique “active” evasion mechanisms to achieve a prolonged circulation time in bloodstream. For example, the glycoprotein gp42 of the Epstein-Barr virus interacts with MHC-II molecules, where it hinders antigen presentation to CD4+ T-cells [42]. In the case of HIV-1, the envelope protein gp120 binds to CD4 molecules and inhibits the interaction of CD4 and MHC-II [43]. The envelope protein E2 of hepatitis C virus (HCV) binds to CD81, which prevents natural killer (NK) cell-mediated lysis and cytokine release [44].
Viruses can also be shielded by an envelope layer consisting of highly glycosylated proteins. The barrier can reduce recognition by the immune system [45]. This strategy operates early in the infection of a host cell, following virion secretion. Lipid and protein derived from the host cell are utilized. The strategy operates in the evasion of the complement system, a major non-specific host defense mechanism [45]. Cowpox virus is equipped with an inhibitor of complement (i.e., the inflammation modulatory protein), which inhibit the production of C3a and C5a during the infection [46]. Viruses including human cytomegalovirus and vaccinia virus utilize similar strategy, by incorporating host CD59 proteins in to the envelope, resulting in the protection from complement lysis [20,47]. The complement system consisting of the classic, alternative, and lectin pathways, is also important in the immune response [48], which recognizes invading pathogens and activates antiviral activities [49]. However, some viruses can evade these complement systems. For example, the HIV-1 envelope proteins (gp120 and gp41) [50] and the West Nile virus NS1 [51] bind to factor H, which regulates the alternative pathway of complement activation in the bloodstream and on the cell surface [52]. In the case of HIV-1, the amino acid sequence 105-119 of gp41 was indicated to interact with factor H immediately after dissociation of the envelope complex [53].
Viruses may become “invisible” to the host by genetic mutation. HBV and HCV harboring specific mutations in the envelope protein are “stealth mutants”. These mutant viruses can escape from the host immune reaction due to the modification of a viral protein epitope that is recognized by the host B or T cells [54]. Although the PEGylation of nanocarriers, which has been approved by the United States Food and Drug Administration (FDA) as a nanomedicine vehicle [55,56], repetitive administration of PEGylated nanocarriers may elicit anti-PEG antibodies, which leads the rapid clearance of PEGylated nanocarriers from body [57,58]. The aforementioned viral systems shall contribute to the development of stealth DDSs as novel strategy to evade from host defense mechanisms.
2.2. Tissue tropism
Virus targeting is a rigorous and complicated step leading to successful endocytosis and infection. Generally, viruses first interact with one or more initial receptor(s) on the target cell surface, which could affect the virus structure and activate some viral surface proteins, initiating other receptor interaction and endocytosis development [59].
2.2.1. Heparan sulfate proteoglycan
Heparan sulfate proteoglycan (HSPG) is a glycoprotein located in the close proximity to cell surface and extracellular matrix, consisting of core protein and covalently attached heparan sulfate chains [60]. Many types of HSPGs are found in cellular membrane, extracellular matrix, and secretory vesicles. Membrane-bound HSPGs contain syndecans and glycosylphosphatidylinositol-anchored proteoglycans (glypicans) [61,62]. HSPGs in the extracellular matrix contain agrin, perlecan, and type XVIII collagen [63]. HSPGs in secretory vesicles contain serglycin [60]. Interaction with HSPG plays many important roles in biological functions, such as cell migration, cell proliferation, cell recognition, and endocytosis. In the early infection of viruses, HSPG plays as an initial and low-affinity receptor that is responsible for cell recognition and may also trigger conformational change in virus envelope protein [60]. Many viruses interact with HSPG in their early infection step, such as dengue virus [64], HIV-1 [65], adeno-associated viruses [66], herpes simplex virus-1 [67], and HBV [68].
2.2.2. Sialic acid
Sialic acid is a derivative of neuraminic acid and ubiquitously expressed in higher vertebrates [69]. It was firstly identified as a low-affinity virus receptor, leading to the cell entry of viruses, such as IFV [70], parainfluenza virus [71], adenovirus (ADV) [72], and coronavirus [73]. In the case of IFV, the hemagglutinin (HA) protein binds sialic acid, initiating the infection by inducing fusion between viral and cellular membranes [74].
2.2.3. Specific high-affinity receptors
Besides low-affinity receptors including HSPGs and sialic acids, each virus generally requires specific high-affinity receptors which determine tissue tropism. Some viruses utilize multiple high-affinity receptors for their successful infection [59]. As an example, human hepatotropic virus HCV is an enveloped RNA virus with a 9.6-kb genome. It belongs to the Hepacivirus genus of the Flaviviridae family [75]. Cell entry of HCV occurs as follows: HCV interacts with low-density-lipoprotein receptor and HSPG on the surface of human hepatocytes [76]. Subsequent interaction with scavenger receptor class B member 1 exposes the CD81-binding domain located at the HCV E2 glycoprotein [77]. HCV binds to CD81 at the tight junctions [44] and then interacts with claudin 1 [78], which induces clathrin-mediated endocytosis [79]. While occludin [80] and Niemann-Pick C1-like 1 [81] were shown as essential receptors, their precise role in the infection process is currently unknown.
2.3. Cell entry
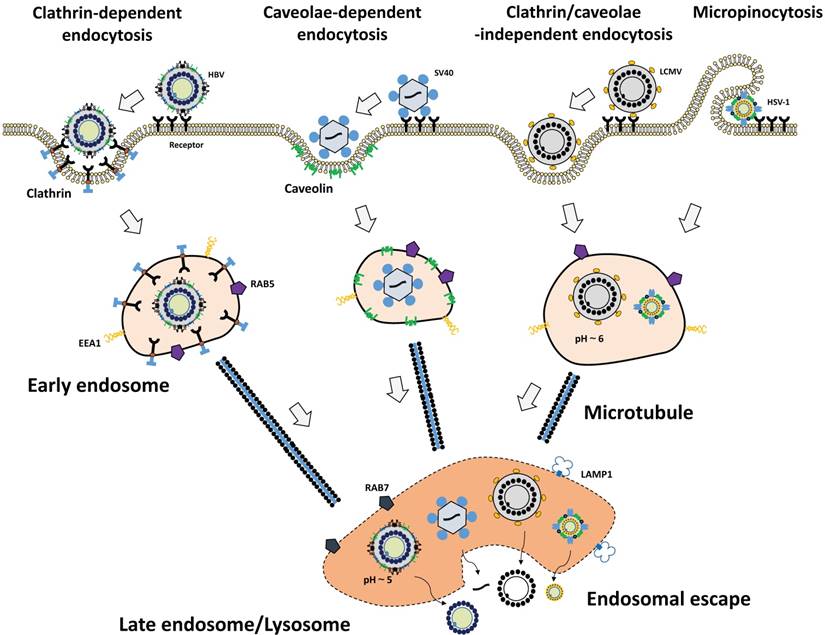
After interaction with viral receptors, a virion can enter the cell efficiently for successful infection. Endocytic vesicles carry viruses from the plasma membrane to the perinuclear area [59]. According to the plasma protein of host cell, most of viruses utilize the endocytosis pathways described below: clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis (Fig. 1). Additionally, several clathrin- and caveolae/raft-independent mechanisms are involved in virus entry [82].
Cellular uptake mechanisms of viruses. Viruses are taken up by cells via various types of endocytosis pathways upon binding to their receptors. HBV (far left) binds to the receptor and clathrin-mediated endocytosis occurs. SV40 (middle left) is taken up by caveolae-dependent endocytosis. Lymphocytic choriomeningitis virus (LCMV) (middle right) enters cells via clathrin/caveolae-independent endocytosis. HSV-1 (far right) is internalized by macropinocytosis. Following the endocytosis, viruses escape from late endosomes or lysosomes, release their contents to cytoplasm, and establish infection in host cells. EEA1, early endosome antigen 1; LAMP1, lysosome-associated membrane protein 1; Rab5, marker for early endosomes; Rab7, marker for late endosomes.
2.3.1. Clathrin-mediated endocytosis
Clathrin-mediated endocytosis is the most extensively studied and best understood endocytosis pathway, including formation of clathrin-coated pits [59] and vesicles, cargo recruitment, and vesicle fission [83]. Clathrin-mediated endocytosis occurs in all cell types and necessary for the turnover of membrane proteins and lipids, uptake of nutrients [83]. Viruses exploit the clathrin-mediated endocytosis process to gain entry to host cells, as shown in Semliki Forest virus (SFV) [84,85], vesicular stomatitis virus (VSV) [86], HCV [87], ADV [88], and dengue virus [89]. The clathrin-mediated endocytosis carries cargo molecules from the plasma membrane to early endosomes within 2 min. [59]. Clathrin-mediated endocytosis of viruses is generally quick. In the case of dengue virus, the virus particle firstly moves along the plasma membrane for about 2 minutes before associating with clathrin-containing domains. Next, the virus is delivered to the late endosomes within 5.5 min after cell entry, and fused with endosomal membrane within 12.5 min [59,89]. Following the clathrin-mediated endocytosis, the virus cargo is transported to the early endosome within several minutes by the help of Rab5 protein. Some viruses immediately move to late endosome by a switch of Rab subsets [90] and start to fuse with endosomal membrane in acidic condition within several minutes [79].
2.3.2. Caveolae-mediated endocytosis
Caveolae-dependent endocytosis begins with the formation of endocytic vesicles containing cholesterol and lipid rafts [91]. After cell entry, the cargo was transferred to early endosomes, late endosomes, and endoplasmic reticulum. For instance, SV40 uses these pathways through binding to glycosphingolipids as their receptors [82].
2.3.3. Macropinocytosis
Macropinocytosis is an actin-dependent endocytic event, leading to internalization of cargo into a large vesicle. The activation of actin and microfilaments connected to the plasma membrane can induce ruffling of the plasma membrane, leading to macropinocytosis [92]. Vaccinia virus [93], ADV [88], and other virus families utilize micropinocytosis. During the internalization, virus particles first induce the actin-mediated membrane ruffling and blebbing. Then, the large vesicles called macropinosomes are formed at the plasma membrane. Macropinosomes internalize viruses and the translocation of viruses to the cytoplasm is mediated through the limiting membrane of the macropinosomes [94].
2.4. Endosomal escape
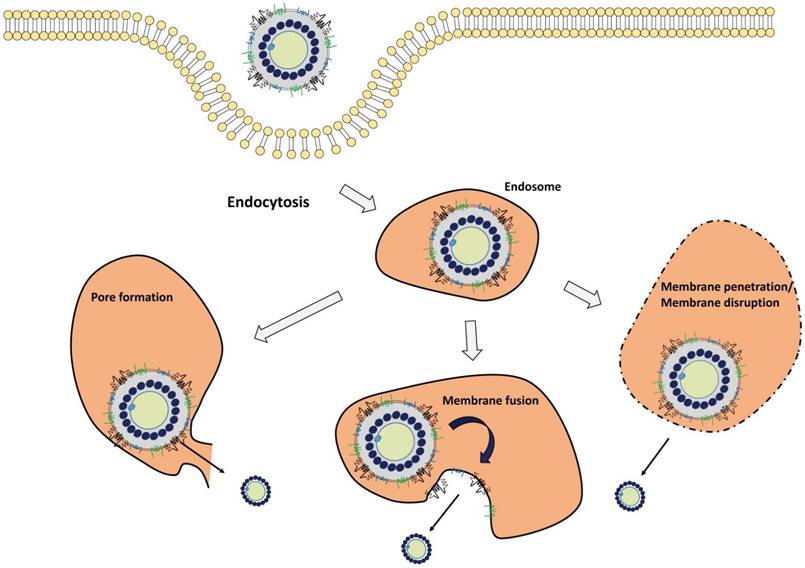
Successful infection of virus requires surmounting the barrier imposed by the cellular membrane, especially the endosome/lysosome membrane. The same problem faced to virus-inspired nanomedicine in the delivery of the therapeutic payload to the cytoplasm. Here, we summarize three pathways of endosomal escape of viruses: membrane fusion, membrane pore formation, and membrane penetration (Fig. 2).
Endosomal escape mechanism of viruses. Endocytosed viruses are sorted to the early endosome. With endosome maturation, the pH of endocytic vesicles decreases. Depending on the surrounding environment, viruses are activated to exert endosomal-escaping functions such as membrane pore formation, membrane fusion, membrane penetration, and membrane disruption.
2.4.1. Membrane fusion
Membrane fusion is a major strategy in the endosomal escape of enveloped virus for their uncoating, leading to the release of viral components that include viral genome, capsids, and polymerase into the cytoplasm [95]. Generally, the fusogenic domain of envelope proteins is responsible for this process. Under the acidic conditions in late endosome, the fusion protein undergoes a conformational change, leading to the insertion of hydrophobic fusogenic domain into the endosomal membrane to form a dehydration interface and hemifusion stalk. In case of IFV, the HA protein undergoes a conformational change after protonation at acidic pH, leading to the fusion peptide with an exposed helical structure, which results in the fusion between viral and endosomal membranes [96]. This membrane fusion mechanism was found in other viral envelope proteins including SFV E1 protein [97] and HIV-1 gp41 protein [98]. Although low pH is important for the structural change of viral proteins, some viruses have different suitable conditions. For example, SFV and VSV fuse with early endosomal membrane at relatively high pH (~pH 6), whereas IFV typically fuses with late endosome membrane at lower pH (~pH 5).
2.4.2. Membrane pore formation
Membrane pore formation in the endosome is another strategy for efficient endosomal escape of viruses, especially non-enveloped viruses. Two models are proposed to form pores in the lipid bilayer: barrel-stave pores [99] and toroidal channels [100]. In the barrel-stave pore model, peptides reorient to become the staves that collectively form a barrel-shaped cluster in a perpendicularly oriented manner to the plane of lipid bilayer, resulting in the formation of a pore [99]. In the toroidal channel model, peptides aggregate, enter the membrane in a perpendicular orientation, and then curve inward to form a pore lined by the peptides [100]. In the case of ADV, the penton base protein, a hydrophobic viral protein, is responsible for pore formation in the acidic environment [101]. Rhinovirus [102], poliovirus [103], and coxsackievirus [104] are also involved in this strategy of pore formation.
2.4.3. Membrane penetration
HIV-1 tat protein is a powerful transcription factor of the integrated HIV-1 genome, which has been utilized in the virus-inspired nanomedicine application to enhance endosomal release [105]. The positively charged Arg and Lys residues of tat protein interact with the negatively charged membrane, leading to lipid membrane penetration, which facilitates gene or drug delivery via membrane destabilization [106]. Other peptides, such as human papillomavirus L2 peptide also exhibits membrane-destabilizing activity under acidic conditions [107].
3. Virus-inspired nanocarriers
Ideal nanocarrier should harbor virus-inspired abilities as mentioned above: immune system evasion, tissue tropism, cell entry, and endosomal escape. Since conventional nanocarriers have not yet fully equipped with these abilities, various virus-inspired nanocarriers have been developed so far by mimicking viral infection machineries. In this review, we have divided these nanocarriers into two major groups: viral components-based nanocarriers and non-viral components-based nanocarriers.
3.1. Viral components-based nanocarriers
Nanocarriers with the viral infection machinery are often composed of virus-derived peptide or proteins. These viral components enhance the binding and uptake into target cells, followed by release of the payload into the cytoplasm. Virus-like particles (VLPs) are composed of recombinant viral proteins. When the structural proteins of viruses are expressed in exogenous cells, recombinant VLPs can be formed like the native virus. In other cases, purified capsid proteins can form VLPs in vitro in a self-organizing manner. Other types of nanocarriers are hybrids of viral components and synthetic materials.
3.1.1. Nanocarriers with viral components
The simplest way to endow synthetic nanocarriers with virus-derived infection machinery is conjugation of viral functional components. The aforementioned infection machineries involved in tropism, cell entry, and endosomal escape are mainly mediated by viral proteins. The virus tropism could be simply transplanted to synthetic nanocarriers by the conjugation of the host cell receptor-recognition domain of viral protein. The synthetic peptide corresponding to the N-terminal region of the HBV envelope L protein (e.g., myristoylated pre-S1(2-47) peptide) interacts with the high-affinity HBV receptor, sodium taurocholate cotransporting polypeptide (NTCP) [108]. When this peptide is conjugated with synthetic nanocarriers, such as LPs [109], and lipoplex (complex of cationic LPs and plasmid DNA) [110], they can target human hepatic cells specifically. The tropism of HBV is strictly limited to human hepatocyte probably due to the restricted expression of the HBV receptor NTCP in hepatocytes [108]. Thus, these HBV-mimicking strategies are expected as highly specific to liver and therefore may reduce the side-effect in non-liver organs.
Envelope spike G glycoprotein of VSV (VSV-G) is frequently used to enhance cellular uptake of the recombinant virus. Since VSV-G contains a fusogenic peptide [111], it could enhance membrane fusion and subsequent cytosolic delivery of drugs. When purified VSV-G are mixed with lipoplex, the transfection efficiency of lipoplex is strongly enhanced [112]. This effect is mediated by the fusogenic activity of VSV-G, as shown by the loss of transfection efficiency of the lipoplex in a fusion-impaired mutant VSV-G. Similar to VSV-G, HA protein of IFV could enhance gene transfer. When HA-2 protein is conjugated to the complex formed with plasmid DNA and transferrin-conjugated poly-Lys, the transfection efficiency is significantly enhanced [113]. Thus, viral components are useful to endow synthetic nanocarriers with the function derived from viruses for efficient drug delivery.
The virosome is a complex of LPs and viral components, another type of virus-inspired nanocarrier [114]. The concept of the virosome was substantiated in 1975 in an experiment where LPs and subunit proteins from IFV were mixed [115]. They succeeded in the efficient transfer of plasmid DNA in vitro and in vivo by using virosomes constructed from Sendai virus [116,117]. Virosomes can be modified with targeting ligands. For example, virosome constructed from IFV HA-2 protein was modified with anti-HER2 Fab fragments [118]. After the encapsulation of the anti-cancer drug doxorubicin, Fab-modified virosomes can deliver drugs by targeting HER-2, and then kill tumor cells in vitro and in vivo. Virosomes with IFV proteins have been used for the delivery of antigens to elicit immune reaction in dendritic cells in vitro [119]. The virosome can also be an efficient delivery platform for protein vaccine.
One of the advantages of these types of nanocarriers is that a part of viral components can be chemically synthesized. For instance, functional domain of viral proteins can be synthesized as a short peptide. Moreover, viral envelope membrane can be substituted by conventional LPs. Thus, these simplified virus-like nanocarriers can resembles the infection machinery of parental viruses.
3.1.2. VLPs
Various types of structural proteins of virus are known to spontaneously form VLPs when viral proteins are expressed in exogenous cells. In the case of enveloped viruses, viral proteins require a lipid bilayer from the host cell membrane for the assembly of VLPs. As VLPs are fully composed of viral components, they might functionally resemble the parental viruses. To date, various types of VLPs from a broad range of virus have been studied for drug delivery applications [120]. VLPs of human papilloma virus were utilized for the delivery of plasmid DNA [121]. After the intramuscular injection of papilloma virus VLPs with plasmid DNA in mice, plasmid DNA was successfully delivered to antigen presenting cells and gene expression was induced. This platform is useful for the delivery of DNA vaccine to elicit a functional immune reaction against pathogens. Furthermore, VLPs can be modified to deliver protein cargos by fusing the components of VLPs and therapeutic proteins. In a study of engineered VLPs of SV40, minor SV40 VP2/3 proteins were fused with model proteins (EGFP or yeast cytosine deaminase) and incorporated into VLPs composed of major VP1 proteins. The engineered VLPs could functionally deliver the encapsulated proteins into cells [118]. Interestingly, VLPs may resemble the biodistribution of parental viruses upon the administration into the host. For example, oral administration of rotavirus VLPs is followed by their penetration to ileum and colon [122]. These VLPs are of interest for the delivery of drugs to the gastrointestinal tract.
As mentioned above, VLPs can fully resemble the infection machinery of parental viruses. Although VLPs are promising platform as DDS, VLPs may elicit immunoreaction in human due to the immunogenic epitope of viral proteins. For the clinical application, immunogenic epitopes should be modified to decrease the immunogenicity of VLPs.
3.1.3. Complexes of synthetic materials and viruses
Some researchers have tried to establish novel class of delivery material by combining virus and synthetic materials [123]. This approach could endow viruses with additional functions and properties. The hybrid of baculovirus and cationic polymer polyethyleneimine has been utilized for gene delivery [124]. By utilizing functional synthetic materials, viruses can be endowed with unique characteristics. For instance, cowpea chlorotic mottle virus was fabricated with temperature-switchable polymers [125]. This hybrid nanocarrier showed temperature-dependent assembly/disassembly properties, indicating that virus can modify synthetic materials to respond to environmental factors, such as temperature and pH.
Although these strategies are promising to endow viruses with additional functions, intrinsic immunogenicity and safety issues of viruses remains.
3.2. Non-viral component-based nanocarriers
The structure of viruses is tightly linked to their biological function. Viruses protect their genome in the inner space of a virion from exogenous degradation. Virions display surface proteins that attach with the host cell membrane. The outer envelope structure interacts with host cell membrane/receptors and finally releases the inner core, followed by the delivery of the genome into the host nucleus. This structural hierarchy might be a rational strategy to generate synthetic delivery system for the intracellular drug delivery. The structure of viruses inspired us to construct a synthetic nanocarrier with virus-like “core-shell” structure by fabricating non-viral components, such as nucleic acids, proteins, polymers, and LPs [126]. Nucleic acids are firstly condensed with cationic proteins or polymers, and then coated with other materials. Glover et al. succeeded in generating virus-inspired NPs by combining plasmid DNA and designed proteins [127]. These materials functioned in DNA compaction (protamine), endosomal escape (diphtheria toxin), and targeting (alpha-melanocyte-stimulating hormone). These proteins can interact with plasmid DNA and then form a 70-nm diameter NP capable of delivering plasmid DNA to achieve enhanced transfection efficiency in non-dividing cells. Other group established a multifunctional envelope-type nanodevice (MEND) using plasmid DNA, poly-Lys (DNA compaction), lipids (shell), and stearated octa-arginine peptide (targeting and membrane penetration) [128]. Owing to the strong cell penetrating activity of octa-arginine, MEND can efficiently transfer plasmid DNA into cells in vitro.
Other than enveloped viruses, capsid viruses are alternatives for the nanocarriers harboring virus-inspired structures. ADV is one of the most common viruses for the delivery of foreign genes into cells in both experimental and clinical settings [129]. ADV utilizes integrins on cell surface for the cell entry [130]. Since ADV interacts with integrins by repeated RGD (Arg-Gly-Asp) motifs, Erbacher et al. found that RGD-conjugated cationic polymer (polyethyleneimine) can resemble the early infection machinery of ADV [131]. Plasmid DNA was condensed by mixing with polyethyleneimine derivatives via electrostatic interaction. The resulting complexes exhibited RGD-dependent strong transfection efficiency in vitro.
Protein nanocages can be applied for the virus-inspired DDS. It is well-known that proteins can form nanostructure by self-assembly. Ferritin, heat shock proteins, and vault proteins were shown to form protein nanocages with 10 to 100 nm in diameter and used for DDS applications [132,133]. By the combination of these protein nanocages and viral components, they may resemble the infection machinery of viruses. Murata et al. succeeded in the targeting of ferritin nanocages to human hepatocytes by fusing with hepatocyte-binding peptide derived from HBV [134].
Virus-like structure made by pure chemical components are of interesting. For instance, assembly of polymer chain into virus-like nanostructure is considered as virus-inspired DDS nanocarriers [135]. When the polymer chain contains both hydrophilic and hydrophobic domains, they can be assembled into the core-shell structure with 10 to 100 nm. After the encapsulation of payloads and modification with targeting moieties, the polymeric nanoparticles act as DDS nanocarriers [4]. Other approach includes the self-assembly of microparticles into virus-like structure. Evers et al. reported that synthetic colloidal particles functionalized with chemical groups can form virus-like microcapsule [136]. These results suggested that the pure chemical materials can be utilized for the virus-inspired DDS even though the components are completely distinct from biomolecules.
4. Bio-nanocapsule
4.1. HBV and bio-nanocapsule
Our group has been working on the development of HBV-inspired nanocarriers (designated as bio-nanocapsule (BNC)) for the last two decades. BNC is composed of HBV envelope L proteins embedded in LP, harboring stealth activity, human liver-specific targeting activity, cell entering activity, and endosomal escaping activity. Here, we describe the structure of BNC, infection machinery of HBV/BNC, retargeting of BNC, and future perspectives of BNC.
4.1.1. Generation of BNC
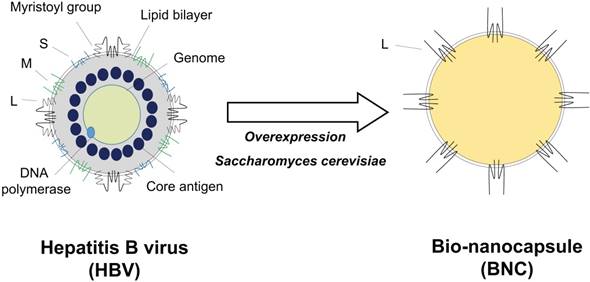
A considerable proportion of the population in the world suffers from HBV infection. Since no effective anti-HBV drug is available, anti-HBV vaccination is important for the protection from infection. Therefore, recombinant HBV subviral particles have been produced since 80s by using eukaryotic cells as immunogens of HB vaccine. Since conventional HB vaccine is composed of HBV envelope S proteins and a LP (also known as hepatitis B surface antigen, HBsAg), more than 5% of vaccinees have never respond to HB vaccine (i.e., non-/low-responders). For enhancing the antigenicity and immunogenicity of HB vaccine as much as possible, we tried to express a complete envelope protein L (pre-S1 + pre-S2 + S region) as particles in recombinant yeast cells (Fig. 3, left), which was expected to elicit additional HBV-neutralizing antibodies. While many attempts had been made by other researchers to synthesize L particles, the N-terminal sequence of the pre-S1 region strongly inhibited its synthesis. We finally overcame the effect by the N-terminal fusion of a chicken lysozyme-derived signal peptide. We succeeded in the overexpression of L particles in yeast cells (approximately 40% of the total soluble protein) [137]. The particles were purified by heating, affinity chromatography, and size exclusion chromatography [138]. Hollow and spherical particles with approximately 100 nm in diameter were obtained. One L particle was estimated to contain about 110 L proteins and an endoplasmic reticulum membrane-derived lipid bilayer. The surface localization of the pre-S1 region, pre-S2 region, and a portion of the S region (antigenic loop, AGL) on the L particles was shown to be similar to HBV. Since surface structure of L particles is similar to that of HBV, we utilized L particles as an HBV-inspired nanocarrier. We designated the L particle as bio-nanocapsule (BNC, Fig. 3 right).
Structure comparison of HBV and BNC. HBV envelope is composed of a lipid bilayer and three types of envelope proteins (S, M, and L). HBV virion contains core proteins, DNA genome, DNA polymerase, and protein X (right). BNC is a hollow nanoparticle produced by yeast cells. BNC is composed of a lipid bilayer derived from the yeast endoplasmic reticulum and HBV envelope L proteins.
4.1.2. Mimicking the early infection machinery of HBV
Though several decades have passed from the identification of HBV in HB patients, the infection mechanism of HBV has been largely unknown. In 2012, NTCP (see chapter 2.2.3.) was at last identified as an essential high-affinity HBV receptor, interacting with the N-terminal myristoylated pre-S1 domain of HBV [108]. Since the exogenous expression of NTCP was shown to confer infection susceptibility for HBV in non-susceptible cells and the expression of NTCP is limited in hepatocytes, the hepatotropism of HBV could be well-explained by the existence of NTCP in hepatocytes. In addition to NTCP, HSPG (see chapter 2.2.1.) plays a crucial role in the early infection steps of HBV. Presumably, one of HSPG, glypican 5, attaches to the HBV surface for cell entry [139]. Conserved residues in the AGL of S region (see above) could interact with HSPG [140,141], possibly leading to the conformational changes in L protein and facilitating the binding of pre-S1 region to other receptors like NTCP.
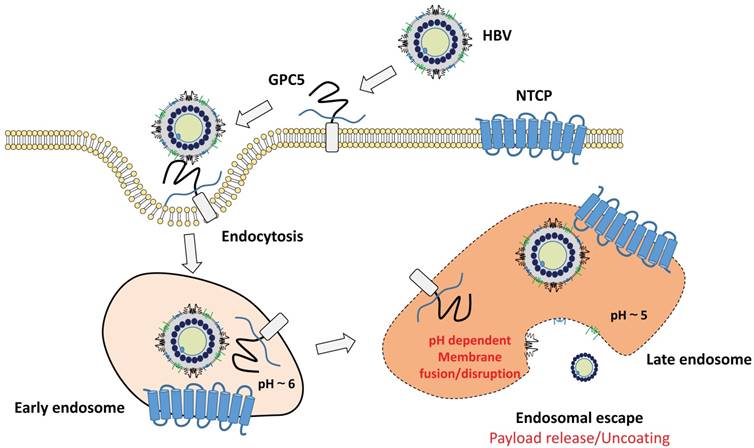
As described above, BNC and HBV share with the similar outer structure, and hence BNC was expected to enter hepatocytes. Recently, we identified the cell entry of BNC is mediated by clathrin-mediated endocytosis and macropinocytosis, as well as patient-derived HBsAg particles in human hepatic cells, of which the cell entry speed was almost the same as that of HBV [142]. When BNC was chemically conjugated with myristoyl group, essential for the HBV infectivity [143], the Myr-BNC could block the HBV infection in vitro competitively, whereas BNCs slightly [144]. Thus, Myr-BNC was shown to utilize the infection machinery of HBV fully. When NTCP was overexpressed in human hepatic cells, we unexpectedly observed no enhancement of cellular uptake of Myr-BNCs, suggesting that NTCP on cell surface could not contribute to the binding with Myr-BNC as well as HBV [144]. These observations led us to propose the refined model of the infection mechanism of HBV (Fig. 4). On the surface of human hepatic cells, HSPG (presumably glypican 5 [139]) may be the main low-affinity receptor for the endocytosis of HBV. Upon endosome maturation, HBV is more likely to bind to NTCP and release their genome into the cytoplasm upon membrane fusion [145]. Further details have yet to be determined and await further studies that will clarify how these receptors coordinately work as HBV receptors.
Proposed model of early infection machinery of HBV. At the cell surface, HBV binds to HSPG, presumably glypican 5 (GPC5), and is endocytosed into cells. NTCP may not be involved in the initial endocytosis step of HBV. After endocytosis, HBV may interact with NTCP upon endosome maturation. The role of NTCP during intracellular trafficking of HBV is unknown. However, the myristoylated N-terminal region of L protein would interact with NTCP in the endocytic vesicles. The binding of HBV and NTCP should be critical for the subsequent infection steps, including membrane fusion and endosomal escape of HBV.
4.2. BNC as HBV-inspired DDS nanocarrier
4.2.1. Stealth activity
NPs injected in the bloodstream should overcome RES for longer circulating time. It was previously demonstrated that administration of a single HBV virion can establish infection of hepatocytes in chimpanzees in every case [146]. This means that the single HBV virion escaped from RES, likely owing to the endogenous stealth activity of HBV. As described above, viruses have many strategies to evade the immune system with viral protein being key (see chapter 2.1.). Since HBV has polymerized-albumin receptor (PAR) in the pre-S2 region, HBV can bind to polymerized human serum albumin (HSA) [147]. When NPs are conjugated with peptide containing PAR, the phagocytosis of NPs by Kupffer cells was reduced (unpublished data). Thus, the recruitment of blood-derived HSA to the PAR domain of HBV (i.e., albumin-coating strategy) may confer evasive abilities on BNCs.
HBV could escape from the host immune reaction by introducing mutations in envelope proteins [148, 149]. When these escape mutant sequences were incorporated into the L protein of BNC, the induction of anti-BNC antibodies was impaired in mice upon repetitive immunization (4 weeks-intervals, for 28 weeks) (unpublished data). This strategy might be applicable for human use of BNC nanocarriers. Naturally occurring escape mutant viruses may be a promising platform to establish stealth nanocarriers that evade the host immune reaction.
Seitz et al. reported the dynamic structural change of the pre-S1 region of HBV occurs by the in vitro incubation at 37°C for several hours [150]. Before incubation, secreted HBV could not bind to HSPG (N-form HBV), but after several hours at 37°C, HBV became to bind to HSPG (B-form HBV). Additionally, N-form HBV was non-infectious in the in vitro infection experiment, while B-form HBV was highly infectious. However, in the in vivo infection experiment using a mouse xenograft model, the N-form HBV could infect human liver cells, while B-form HBV could not. These results suggested that the HSPG binding is necessary for the infectivity of HBV in vitro, but the HSPG binding allows non-specific accumulation of HBV in non-target tissues and then prevents the liver specific accumulation of HBV. During the blood circulation, HBV may change its structure from N-form to B-form. This unique property of HBV has inspired us to establish nanocarriers that can convert the stealth form to the active form during blood circulation. Further study regarding stealth activity of HBV in vivo will help the generation of a novel class of stealth nanocarriers.
4.2.2. Human hepatic cell-specific targeting
The tropism of HBV is strictly limited to human hepatocytes. Therefore, BNCs were expected to exhibit HBV-mimicking infection machineries in human hepatocytes. When the fluorophores or plasmids encoding enhanced green fluorescence protein were incorporated by electroporation, BNCs can deliver them to human hepatic cells specifically in vitro [151]. After the intravenous injection, BNCs were accumulated in the human hepatic cell-derived tumor in xenograft mice [151] or normal human liver tissues transplanted in severe combined immunodeficiency mice [152]. These results indicated that BNCs can target human hepatic cells in vitro and in vivo by infection machinery of HBV.
BNCs can fuse with LPs and form BNC-LP complexes [153,154]. Furthermore, high temperature and low pH facilitates the fusion between BNCs and LPs, leading to the formation of BNC-virosomes [155]. When BNC-virosomes containing doxorubicin were injected into hepatic tumor-bearing xenograft mice, the growth of hepatic cell-derived tumors was inhibited effectively and specifically, strongly suggesting that BNC-virosomes deliver their payload into tumor efficiently utilizing HBV-derived infection machinery [155]. Furthermore, BNC-virosomes were found to deliver payloads into the cytoplasm [145]. These results indicated that virosomes are promising cytoplasmic nanocarriers in drug/gene delivery.
4.2.3. Retargeting of BNC
For expanding the possibility of BNC, we have tried to retarget BNC from human hepatic cells/liver to other cells/tissues for the treatment of various diseases. First, part of pre-S1 region of L protein was substituted for epidermal growth factor (EGF) by genetic modification [151]. This EGF-displaying BNC can target EGFR-overexpressing cells in vitro. Other retargeting strategies of BNC were summarized in a previous review [156]. To date, we have succeeded in the retargeting of BNC to various cells and tissues, constituting 26 in vitro and 10 in vivo delivery experiments. Hence, BNCs could achieve therapeutic effects in non-human liver tissues.
4.2.4. Endosomal escape of BNC
As described in chapter 2.3., uncoating and endosomal escape is necessary for envelope viruses to deliver viral genome into cytoplasm. HBV was postulated to escapes from endosomes by membrane fusion [157]. Previously, three domains have been proposed as fusogenic domain of HBV, including N-terminal part of S region [157], C-terminal half of pre-S2 region [158], and the whole pre-S1 region [159]. Nevertheless, it was not fully understood which domain is responsible for the membrane fusion and uncoating of HBV. In 2015, we identified 16-mer low pH-dependent fusogenic domain in the pre-S1 region by using BNCs and peptide-displaying LPs [145]. The peptide corresponds to the Asn-9 to Gly-24 in the pre-S1 region. Furthermore, mutation analysis of this fusogenic peptide revealed that the hydrophobicity of the peptide correlates with its fusogenic activity, and Asp-16 and Asp-20 might be crucial for the low-pH dependent fusogenic activity [160]. Since the fusogenic activity of BNC was inhibited by the pre-incubation with anti-pre-S1 antibody, the fusogenic domain from Asn-9 to Gly-24 is a dominant region over other fusogenic domains reported previously [145].
4.3. Next-generation BNC
As described in the previous review of BNC [156], the NPs for ideal DDS nanocarriers should harbor the following six activities: self-organizing activity, stealth activity, targeting activity, cell entry activity, and endosomal escaping activity. Viruses have evolved to transfer their genetic materials into the nucleus of target cells in vivo, it has thus been considered that viruses are naturally occurring ideal DDS nanocarriers. Whereas most artificial DDS nanocarriers have still been far from the ideal DDS nanocarriers, which harbor a part of the six activities. In this review, we described that BNC is a nearly sole DDS nanocarrier that accomplishes the equipment of all activities. However, for moving to clinical settings, it would be very hard for pharmaceutical companies to develop BNC-based nanomedicines, because the nanomedicines have to be produced under at least two good manufacturing protocol (GMP) guidelines (biologics and chemicals). Thus, these situations have led us to reconstitute the NPs harboring all activities (equal to next-generation BNC) by using chemically defined materials. The concept of next-generation BNC is similar to the non-viral components-based nanocarriers (see chapter 3.2.). For examples, the surface of LPs should be modified with myristoylated pre-S1(2-47) peptide (for targeting and endosomal escaping activity) and pre-S2(120-129) peptide (for stealth activity) to achieve the efficient in vivo delivery and intracellular trafficking in an HBV/BNC-like manner. The weight ratio of these peptides to LPs should be minimized, and the drug encapsulation rate should be maximized. For retargeting of next-generation BNC, the N-terminal part of pre-S1(2-47) peptide (containing NTCP-binding site) should be changed to other targeting molecules (e.g., DNA aptamer, sugar chain, homing peptide, nanobody). Moreover, since nanomicelles and lipid NPs do not require membrane fusion for the endosomal escape of payloads, the fusogenic domain (pre-S1(9-24)) should be replaced with the peptides for membrane pore formation and membrane penetration (see chapters 2.4.2. and 2.4.3.). These strategies for fabricating next-generation BNC might produce ideal DDS nanocarriers, which could deliver drugs and genes in vivo at the same efficiency of parental viruses.
Conclusion
In this review, we first summarized the infection mechanism of various viruses that might be helpful to establish novel virus-inspired DDS nanocarriers. In the following sections, various virus-inspired nanocarriers, including our platform BNCs, were introduced. According to the low delivery efficacy, conventional nanocarrier-based DDS have been rarely commercialized. Ideally, virus-inspired DDS should resemble the infection machinery of virus while the components are fully synthetic. As mentioned above, some of the infection machineries of virus can be substituted by short peptide that can be chemically synthesized. Furthermore, these short peptides might be less immunogenic than full-length viral proteins. By combining such virus-derived short peptides and synthetic nanocarriers, delivery efficacy of DDS can be improved. The virus-inspired strategy may boost the DDS nanocarriers for the clinical applications.
Abbreviations
ADV, adenovirus; BNC, bio-nanocapsule; DDS, drug delivery system; EPR, enhanced permeability and retention; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV-1, human immunodeficiency virus 1; HSPG, heparan sulfate proteoglycan; IFV, influenza virus; LP, liposome; MHC, major histocompatibility complex; NP, nanoparticle; NTCP, sodium taurocholate cotransporting polypeptide; PEG, poly (ethylene glycol); RES, reticuloendothelial system; PAR, polymerized-albumin receptor; SFV, Semliki Forest virus; siRNA, short interfering RNA; VLP, virus-like particle; VSV, vesicular stomatitis virus.
Acknowledgements
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant-in-Aid for Scientific Research (S) 16H06314 to SK), Grant-in-Aid for JSPS Research Fellow (17J08534 to MS), Japan Agency for Medical Research and Development (AMED, 17cm0106214h0002 and 17fk0310105h0001 to SK), "Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials" from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to SK), and Mishima Kaiun Memorial Foundation (to MS).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Strebhardt K, Ullrich A. Paul Ehrlich's Magic Bullet Concept: 100 Years of Progress. Nat Rev Cancer. 2008;8:473-480
2. Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science (80- ). 2004;303:1818-1822
3. Allen TM, Cullis PR. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv Drug Deliv Rev. 2013;65:36-48
4. Cabral H, Kataoka K. Progress of Drug-Loaded Polymeric Micelles into Clinical Studies. J Control Release. 2014;190:465-476
5. Longmire M, Choyke PL, Kobayashi H. Clearance Properties of Nano-Sized Particles and Molecules as Imaging Agents: Considerations and Caveats. Nanomedicine. 2008;3:703-717
6. Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cnacer Chemotherapy: Mechanism of Tumoritropic Accumulatio of Proteins and the Antitumor Agents Smancs. Cancer Res. 1986;46:6387-6392
7. Byrne JD, Betancourt T, Brannon-Peppas L. Active Targeting Schemes for Nanoparticle Systems in Cancer Therapeutics. Adv Drug Deliv Rev. 2008;60:1615-1626
8. Wilhelm S, Tavares AJ, Dai Q. et al. Analysis of Nanoparticle Delivery to Tumours. Nat Rev Mater. 2016;1:1-12
9. Bertrand N, Wu J, Xu X. et al. Cancer Nanotechnology: The Impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv Drug Deliv Rev. 2014;66:2-25
10. Bae YH, Park K. Targeted Drug Delivery to Tumors: Myths, Reality and Possibility. J Control Release. 2011;153:198-205
11. Venditto VJ, Szoka FC. Cancer Nanomedicines: So Many Papers and so Few Drugs!. Adv Drug Deliv Rev. 2013;65:80-88
12. Lares MR, Rossi JJ, Ouellet DL. RNAi and Small Interfering RNAs in Human Disease Therapeutic Applications. Trends Biotechnol. 2010;28:570-579
13. Dowdy SF. Overcoming Cellular Barriers for RNA Therapeutics. Nat Biotechnol. 2017;35:222-229
14. Gilleron J, Querbes W, Zeigerer A. et al. Image-Based Analysis of Lipid Nanoparticle-Mediated siRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat Biotechnol. 2013;31:638-646
15. Wegst UGK, Bai H, Saiz E. et al. Bioinspired Structural Materials. Nat Mater. 2014;14:23-36
16. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G. et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science. 2000;288:669-672
17. Thomas CE, Ehrhardt A, Kay M. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat Rev Genet. 2003;4:346-358
18. Murphy K, Weaver C. Janeway's Immunobiology. Garland Science. 2016
19. Vossen MTM, Westerhout EM, Söderberg-Nauclér C. et al. Viral Immune Evasion: A Masterpiece of Evolution. Immunogenetics. 2002;54:527-542
20. Alcami A, Koszinowski UH. Viral Mechanisms of Immune Evasion. Immunol Today. 2000;21:447-455
21. Douglas T. Viruses: Making Friends with Old Foes. Science (80- ). 2006;312:873-875
22. Paul D, Achouri S, Yoon Y-Z. et al. Phagocytosis Dynamics Depends on Target Shape. Biophys J. 2013;105:1143-1150
23. Champion JA, Mitragotri S. Role of Target Geometry in Phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930-4934
24. Tiollais P, Pourcel C, Dejean A. The Hepatitis B Virus. Nature. 1985;317:489-495
25. Urban S, Bartenschlager R, Kubitz R. et al. Strategies to Inhibit Entry of HBV and HDV into Hepatocytes. Gastroenterology. 2014;147:1-17
26. Rydell GE, Prakash K, Norder H. et al. Hepatitis B Surface Antigen on Subviral Particles Reduces the Neutralizing Effect of Anti-HBs Antibodies on Hepatitis B Viral Particles in Vitro. Virology. 2017;509:67-70
27. Bruckman MA, Randolph LN, VanMeter A. et al. Biodistribution, Pharmacokinetics, and Blood Compatibility of Native and PEGylated Tobacco Mosaic Virus Nano-Rods and -Spheres in Mice. Virology. 2014;449:163-173
28. Atabekov J, Nikitin N, Arkhipenko M. et al. Thermal Transition of Native Tobacco Mosaic Virus and RNA-Free Viral Proteins into Spherical Nanoparticles. J Gen Virol. 2011;92:453-456
29. Huang X, Li L, Liu T. et al. The Shape Effect of Mesoporous Silica Nanoparticles on Biodistribution, Clearance, and Biocompatibility in Vivo. ACS Nano. 2011;5:5390-5399
30. Chithrani BD, Ghazani AA, Chan WCW. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006;6:662-668
31. Arnida Janát-Amsbury MM, Ray A et al. Geometry and Surface Characteristics of Gold Nanoparticles Influence Their Biodistribution and Uptake by Macrophages. Eur J Pharm Biopharm. 2011;77:417-423
32. Huber VC, Lynch JM, Bucher DJ. et al. Fc Receptor-Mediated Phagocytosis Makes a Significant Contribution to Clearance of Influenza Virus Infections. J Immunol. 2001;166:7381-7388
33. Champion JA, Walker A, Mitragotri S. Role of Particle Size in Phagocytosis of Polymeric Microspheres. Pharm Res. 2008;25:1815-1821
34. Ghigo E, Kartenbeck J, Lien P. et al. Ameobal Pathogen Mimivirus Infects Macrophages through Phagocytosis. PLoS Pathog. 2008:4
35. Grove J, Marsh M. The Cell Biology of Receptor-Mediated Virus Entry. J Cell Biol. 2011;195:1071-1082
36. Tabata Y, Ikada Y. Effect of the Size and Surface Charge of Polymer Microspheres on Their Phagocytosis by Macrophage. Biomaterials. 1988;9:356-362
37. Singh P, Prasuhn D, Yeh RM. et al. Bio-Distribution, Toxicity and Pathology of Cowpea Mosaic Virus Nanoparticles in Vivo. J Control Release. 2007;120:41-50
38. Michen B, Graule T. Isoelectric Points of Viruses. J Appl Microbiol. 2010;109:388-397
39. Moghimi SM, Hunter C, Murray JC. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol Rev. 2001;53:283-318
40. Ploegh HL. Viral Strategies of Immune Evasion. Science. 1998;280:248-253
41. Guidotti LG, Chisari FV. To Kill or to Cure: Options in Host Defense against Viral Infection. Curr Opin Immunol. 1996;8:478-483
42. Paludan C, Schmid D, Landthaler M. et al. Endogenous MHC Class II Processing of a Viral Nuclear Antigen after Autophagy. Science. 2005;307:593-596
43. Raposo G, Moore M, Innes D. et al. Human Macrophages Accumulate HIV-1 Particles in MHC II Compartments. Traffic. 2002;3:718-729
44. Pileri P, Uematsu Y, Campagnoli S. et al. Binding of Hepatitis C Virus to CD81. Science. 1998;282:938-941
45. Rudd PM. Glycosylation and the Immune System. Science (80- ). 2001;291:2370-2376
46. Kotwal GJ. Poxviral Mimicry of Complement and Chemokine System Components: What's the End Game? Immunol Today. 2000;21:242-248
47. Spear GT, Lurain NS, Parker CJ. et al. Host Cell-Derived Complement Control Proteins CD55 and CD59 Are Incorporated into the Virions of Two Unrelated Enveloped Viruses. Human T Cell Leukemia/lymphoma Virus Type I (HTLV-I) and Human Cytomegalovirus (HCMV). J Immunol. 1995;155:4376-4381
48. Carroll MC. The Complement System in Regulation of Adaptive Immunity. Nat Immunol. 2004;5:981-986
49. Müller-Eberhard HJ. Molecular Organization and Function of the Complement System. Annu Rev Biochem. 1988;57:321-347
50. Stoiber H, Pintér C, Siccardi AG. et al. Efficient Destruction of Human Immunodeficiency Virus in Human Serum by Inhibiting the Protective Action of Complement Factor H and Decay Accelerating Factor (DAF, CD55). J Exp Med. 1996;183:307-310
51. Chung KM, Liszewski MK, Nybakken G. et al. West Nile Virus Nonstructural Protein NS1 Inhibits Complement Activation by Binding the Regulatory Protein Factor H. Proc Natl Acad Sci U S A. 2006;103:19111-19116
52. Klein RJ, Zeiss C, Chew EY. et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science. 2005;308:385-389
53. Pintér C, Siccardi AG, Lopalco L. et al. HIV Glycoprotein 41 and Complement Factor H Interact with Each Other and Share Functional as Well as Antigenic Homology. AIDS Res Hum Retroviruses. 1995;11:971-980
54. Wieland SF, Chisari FV. Stealth and Cunning: Hepatitis B and Hepatitis C Viruses. J Virol. 2005;79:9369-9380
55. Harris JM, Chess RB. Effect of Pegylation on Pharmaceuticals. Nat Rev Drug Discov. 2003;2:214-221
56. Jokerst J V, Lobovkina T, Zare RN. et al. Nanoparticle PEGylation for Imaging and Therapy. Nanomedicine. 2011;6:715-728
57. Dams ET, Laverman P, Oyen WJ. et al. Accelerated Blood Clearance and Altered Biodistribution of Repeated Injections of Sterically Stabilized Liposomes. J Pharmacol Exp Ther. 2000;292:1071-1079
58. Ichihara M, Shimizu T, Imoto A. et al. Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats. Pharmaceutics. 2011;3:1-11
59. Mercer J, Schelhaas M, Helenius A. Virus Entry by Endocytosis. Annu Rev Biochem. 2010;79:803-833
60. Sarrazin S, Lamanna WC, Esko JD. Heparan Sulfate Proteoglycans. Cold Spring Harb Perspect Biol. 2011;3:1-33
61. Carey DJ. Syndecans: Multifunctional Cell-Surface Co-Receptors. Biochem J. 1997;327( Pt 1):1-16
62. Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224
63. Iozzo R V. Matrix Proteoglycans: From Molecular Design to Cellular Function. Annu Rev Biochem. 1998;67:609-652
64. Hilgard P. Heparan Sulfate Proteoglycans Initiate Dengue Virus Infection of Hepatocytes. Hepatology. 2000;32:1069-1077
65. de Witte L, Bobardt M, Chatterji U. et al. Syndecan-3 Is a Dendritic Cell-Specific Attachment Receptor for HIV-1. Proc Natl Acad Sci U S A. 2007;104:19464-19469
66. Summerford C, Samulski RJ. Membrane-Associated Heparan Sulfate Proteoglycan Is a Receptor for Adeno-Associated Virus Type 2 Virions. J Virol. 1998;72:1438-1445
67. Shukla D, Spear PG. Herpesviruses and Heparan Sulfate: An Intimate Relationship in Aid of Viral Entry. J Clin Invest. 2001;108:503-510
68. Schulze A, Gripon P, Urban S. Hepatitis B Virus Infection Initiates with a Large Surface Protein-Dependent Binding to Heparan Sulfate Proteoglycans. Hepatology. 2007;46:1759-1768
69. Corfield AP, Schauer R. Occurrence of Sialic Acids. 1982;: 5-50.
70. Weis W, Brown JH, Cusack S. et al. Structure of the Influenza Virus Haemagglutinin Complexed with Its Receptor, Sialic Acid. Nature. 1988;333:426-431
71. Amonsen M, Smith DF, Cummings RD. et al. Human Parainfluenza Viruses hPIV1 and hPIV3 Bind Oligosaccharides with alpha2-3-Linked Sialic Acids That Are Distinct from Those Bound by H5 Avian Influenza Virus Hemagglutinin. J Virol. 2007;81:8341-8345
72. Arnberg N, Edlund K, Kidd AH. et al. Adenovirus Type 37 Uses Sialic Acid as a Cellular Receptor. J Virol. 2000;74:42-48
73. Schwegmann-Wessels C, Herrler G. Sialic Acids as Receptor Determinants for Coronaviruses. Glycoconj J. 2006;23:51-58
74. Lear JD, DeGrado WF. Membrane Binding and Conformational Properties of Peptides Representing the NH2 Terminus of Influenza HA-2. J Biol Chem. 1987;262:6500-6505
75. Moradpour D, Penin F, Rice CM. Replication of Hepatitis C Virus. Nat Rev Microbiol. 2007;5:453-463
76. Burlone ME, Budkowska A. Hepatitis C Virus Cell Entry: Role of Lipoproteins and Cellular Receptors. J Gen Virol. 2009;90:1055-1070
77. Scarselli E, Ansuini H, Cerino R. et al. The Human Scavenger Receptor Class B Type I Is a Novel Candidate Receptor for the Hepatitis C Virus. EMBO J. 2002;21:5017-5025
78. Evans MJ, von Hahn T, Tscherne DM. et al. Claudin-1 Is a Hepatitis C Virus Co-Receptor Required for a Late Step in Entry. Nature. 2007;446:801-805
79. Lindenbach BD, Rice CM. The Ins and Outs of Hepatitis C Virus Entry and Assembly. Nat Rev Microbiol. 2013;11:688-700
80. Ploss A, Evans MJ, Gaysinskaya VA. et al. Human Occludin Is a Hepatitis C Virus Entry Factor Required for Infection of Mouse Cells. Nature. 2009;457:882-886
81. Sainz B, Barretto N, Martin DN. et al. Identification of the Niemann-Pick C1-like 1 Cholesterol Absorption Receptor as a New Hepatitis C Virus Entry Factor. Nature medicine. 2012;18:281-285
82. Kirkham M, Parton RG. Clathrin-Independent Endocytosis: New Insights into Caveolae and Non-Caveolar Lipid Raft Carriers. Biochim Biophys Acta. 2005;1745:273-286
83. McMahon HT, Boucrot E. Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nat Rev Mol Cell Biol. 2011;12:517-533
84. Helenius A, Kartenbeck J, Simons K. et al. On the Entry of Semliki Forest Virus into BHK-21 Cells. J Cell Biol. 1980;84:404-420
85. White J, Kartenbeck J, Helenius A. Fusion of Semliki Forest Virus with the Plasma Membrane Can Be Induced by Low pH. J Cell Biol. 1980;87:264-272
86. Sun X, Yau VK, Briggs BJ. et al. Role of Clathrin-Mediated Endocytosis during Vesicular Stomatitis Virus Entry into Host Cells. Virology. 2005;338:53-60
87. Blanchard E, Belouzard S, Goueslain L. et al. Hepatitis C Virus Entry Depends on Clathrin-Mediated Endocytosis. J Virol. 2006;80:6964-6972
88. Meier O, Boucke K, Hammer SV. et al. Adenovirus Triggers Macropinocytosis and Endosomal Leakage Together with Its Clathrin-Mediated Uptake. J Cell Biol. 2002;158:1119-1131
89. Acosta EG, Castilla V. Damonte EB, Functional Entry of Dengue Virus into Aedes Albopictus Mosquito Cells Is Dependent on Clathrin-Mediated Endocytosis. J Gen Virol. 2008;89:474-484
90. Gorvel JP, Chavrier P, Zerial M. et al. rab5 Controls Early Endosome Fusion in Vitro. Cell. 1991;64:915-925
91. Nabi IR, Le PU. Caveolae/raft-Dependent Endocytosis. J Cell Biol. 2003;161:673-677
92. Swanson JA, Biology C, Avenue L. et al. Macropinocytosis. Trends Cell Biol. 1995;5:424-428
93. Mercer J, Helenius A. Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science. 2008;320:531-535
94. Mercer J, Helenius A. Virus Entry by Macropinocytosis. Nat Cell Biol. 2009;11:510-520
95. White JM, Whittaker GR. Fusion of Enveloped Viruses in Endosomes. Traffic. 2016;17:593-614
96. Skehel JJ, Wiley DC. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu Rev Biochem. 2000;69:531-569
97. Lescar J, Roussel A, Wien MW. et al. The Fusion Glycoprotein Shell of Semliki Forest Virus: An Icosahedral Assembly Primed for Fusogenic Activation at Endosomal pH. Cell. 2001;105:137-148
98. Chan DC, Fass D, Berger JM. et al. Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell. 1997;89:263-273
99. Yang L, Harroun TA, Weiss TM. et al. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys J. 2001;81:1475-1485
100. Hancock REW, Sahl H-G. Antimicrobial and Host-Defense Peptides as New Anti-Infective Therapeutic Strategies. Nat Biotechnol. 2006;24:1551-1557
101. Wiethoff CM, Wodrich H, Gerace L. et al. Adenovirus Protein VI Mediates Membrane Disruption Following Capsid Disassembly. J Virol. 2005;79:1992-2000
102. Zauner W, Blaas D, Kuechler E. et al. Rhinovirus-Mediated Endosomal Release of Transfection Complexes. J Virol. 1995;69:1085-1092
103. Suhy DA, Giddings TH, Kirkegaard K. Remodeling the Endoplasmic Reticulum by Poliovirus Infection and by Individual Viral Proteins: An Autophagy-like Origin for Virus-Induced Vesicles. J Virol. 2000;74:8953-8965
104. van Kuppeveld FJ, Hoenderop JG, Smeets RL. et al. Coxsackievirus Protein 2B Modifies Endoplasmic Reticulum Membrane and Plasma Membrane Permeability and Facilitates Virus Release. EMBO J. 1997;16:3519-3532
105. Torchilin VP, Rammohan R, Weissig V. et al. TAT Peptide on the Surface of Liposomes Affords Their Efficient Intracellular Delivery Even at Low Temperature and in the Presence of Metabolic Inhibitors. Proc Natl Acad Sci U S A. 2001;98:8786-8791
106. Trabulo S, Cardoso AL, Mano M. et al. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals. 2010;3:961-993
107. Kamper N, Day PM, Nowak T. et al. A Membrane-Destabilizing Peptide in Capsid Protein L2 Is Required for Egress of Papillomavirus Genomes from Endosomes. J Virol. 2006;80:759-768
108. Yan H, Zhong G, Xu G. et al. Sodium Taurocholate Cotransporting Polypeptide Is a Functional Receptor for Human Hepatitis B and D Virus. Elife. 2012;1:e00049
109. Zhang X, Zhang Q, Peng Q. et al. Hepatitis B Virus preS1-Derived Lipopeptide Functionalized Liposomes for Targeting of Hepatic Cells. Biomaterials. 2014;35:6130-6141
110. Wang Z, Yuan Z, Jin L. Gene Delivery into Hepatocytes with the PreS/liposome/DNA System. Biotechnol J. 2008;3:1286-1295
111. Roche S, Rey FA, Gaudin Y. et al. Structure of the Prefusion Form of the Vesicular Stomatitis Virus Glycoprotein G. Science (80- ). 2007;315:843-848
112. Abe A, Miyanohara A, Friedmann T. Enhanced Gene Transfer with Fusogenic Liposomes Containing Vesicular Stomatitis Virus G Glycoprotein. J Virol. 1998;72:6159-6163
113. Wagner E, Plank C, Zatloukal K. et al. Influenza Virus Hemagglutinin HA-2 N-Terminal Fusogenic Peptides Augment Gene Transfer by Transferrin-Polylysine-DNA Complexes: Toward a Synthetic Virus-like Gene-Transfer Vehicle. Proc Natl Acad Sci U S A. 1992;89:7934-7938
114. Kaneda Y. Virosomes: Evolution of the Liposome as a Targeted Drug Delivery System. Adv Drug Deliv Rev. 2000;43:197-205
115. Almeida JD, Edwards DC, Brand CM. et al. Formation of Virosomes from Influenza Subunits and Liposomes. Lancet. 1975;2:899-901
116. Kaneda Y, Iwai K, Uchida T. Increased Expression of DNA Cointroduced with Nuclear Protein in Adult Rat Liver. Science (80- ). 1989;243:375-378
117. Kaneda Y, Nakajima T, Nishikawa T. et al. Hemagglutinating Virus of Japan (HVJ) Envelope Vector as a Versatile Gene Delivery System. Mol Ther. 2002;6:219-226
118. Waelti E, Wegmann N, Schwaninger R. et al. Targeting Her-2/neu with Antirat Neu Virosomes for Cancer Therapy. Cancer Res. 2002;62:437-444
119. Bungener L, Serre K, Bijl L. et al. Virosome-Mediated Delivery of Protein Antigens to Dendritic Cells. Vaccine. 2002;20:2287-2295
120. Ludwig C, Wagner R. Virus-like Particles-Universal Molecular Toolboxes. Curr Opin Biotechnol. 2007;18:537-545
121. Malboeuf CM, Simon DAL, Lee YEE. et al. Human Papillomavirus-like Particles Mediate Functional Delivery of Plasmid DNA to Antigen Presenting Cells in Vivo. Vaccine. 2007
122. Trugnan G, Cortes-Perez NG, Sapin C. et al. Rotavirus-like Particles: A Novel Nanocarrier for the Gut. J Biomed Biotechnol. 2010;2010:317545
123. Boeckle S, Wagner E. Optimizing Targeted Gene Delivery: Chemical Modification of Viral Vectors and Synthesis of Artificial Virus Vector Systems. AAPS J. 2006;8:E731-42
124. Kim Y-K, JAE YOUNG C, JIANG H-L. et al. Hybrid of Baculovirus and Galactosylated PEI for Efficient Gene Carrier. Virology. 2009;387:89-97
125. Kostiainen MA, Pietsch C, Hoogenboom R. et al. Temperature-Switchable Assembly of Supramolecular Virus-Polymer Complexes. Adv Funct Mater. 2011;21:2012-2019
126. Mastrobattista E, van der Aa M a EM, Hennink WE. et al. Artificial Viruses: A Nanotechnological Approach to Gene Delivery. Nat Rev Discov. 2006;5:115-121
127. Glover DJ, Ng SM, Mechler A. et al. Multifunctional Protein Nanocarriers for Targeted Nuclear Gene Delivery in Nondividing Cells. FASEB J. 2009;23:2996-3006
128. Kogure K, Moriguchi R, Sasaki K. et al. Development of a Non-Viral Multifunctional Envelope-Type Nano Device by a Novel Lipid Film Hydration Method. J Control Release. 2004;98:317-323
129. Edelstein ML, Abedi MR, Wixon J. et al. Gene Therapy Clinical Trials Worldwide 1989-2004 - An Overview. J Gene Med. 2004;6:597-602
130. Nemerow GR, Stewart PL. Role of Alpha(v) Integrins in Adenovirus Cell Entry and Gene Delivery. Microbiol Mol Biol Rev. 1999;63:725-734
131. Erbacher P, Remy J-S, Behr J-P. Gene Transfer with Synthetic Virus-like Particles via the Integrin-Mediated Endocytosis Pathway. Gene Ther. 1999;6:138-145
132. Lim S. Protein Nanocage: The Versalite Molecular Shell. Asia Pacific Biotech. 2013;17:51-53
133. Lee EJ, Lee NK, Kim IS. Bioengineered Protein-Based Nanocage for Drug Delivery. Adv Drug Deliv Rev. 2016;106:157-171
134. Murata M, Piao JS, Narahara S. et al. Expression and Characterization of Myristoylated preS1-Conjugated Nanocages for Targeted Cell Delivery. Protein Expr Purif. 2015;110:52-56
135. Elsabahy M. Wooley KL, Strategies Toward Well-Defined Polymer Nanoparticles Inspired by Nature: Chemistry versus Versatility. J Polym Sci A Polym Chem. 2012;50:1869-1880
136. Evers CHJ, Luiken JA, Bolhuis PG. et al. Self-Assembly of Microcapsules via Colloidal Bond Hybridization and Anisotropy. Nature. 2016;534:364-368
137. Kuroda S, Otaka S, Miyazaki T. et al. Hepatitis B Virus Envelope L Protein Particles: Synthesis and Assembly in Saccharomyces Cerevisiae, Purification and Characterization. J Biol Chem. 1992;267:1953-1961
138. Jung J, Iijima M, Yoshimoto N. et al. Efficient and Rapid Purification of Drug- and Gene-Carrying Bio-Nanocapsules, Hepatitis B Virus Surface Antigen L Particles, from Saccharomyces Cerevisiae. Protein Expr Purif. 2011;78:149-155
139. Verrier ER, Colpitts CC, Bach C. et al. A Targeted Functional RNA Interference Screen Uncovers Glypican 5 as an Entry Factor for Hepatitis B and D Viruses. Hepatology. 2016;63:35-48
140. Jaoude GA, Sureau C. Role of the Antigenic Loop of the Hepatitis B Virus Envelope Proteins in Infectivity of Hepatitis Delta Virus. J Virol. 2005;79:10460-10466
141. Sureau C, Salisse J. A Conformational Heparan Sulfate Binding Site Essential to Infectivity Overlaps with the Conserved Hepatitis B Virus A-Determinant. Hepatology. 2013;57:985-994
142. Yamada M, Oeda A, Jung J. et al. Hepatitis B Virus Envelope L Protein-Derived Bio-Nanocapsules: Mechanisms of Cellular Attachment and Entry into Human Hepatic Cells. J Control Release. 2012;160:322-329
143. Gripon P, Le Seyec J, Rumin S. et al. Myristylation of the Hepatitis B Virus Large Surface Protein Is Essential for Viral Infectivity. Virology. 1995;213:292-299
144. Somiya M, Liu Q, Yoshimoto N. et al. Cellular Uptake of Hepatitis B Virus Envelope L Particles Is Independent of Sodium Taurocholate Cotransporting Polypeptide, but Dependent on Heparan Sulfate Proteoglycan. Virology. 2016;497:23-32
145. Somiya M, Sasaki Y, Matsuzaki T. et al. Intracellular Trafficking of Bio-Nanocapsule-Liposome Complex: Identification of Fusogenic Activity in the Pre-S1 Region of Hepatitis B Virus Surface Antigen L Protein. J Control Release. 2015;212:10-18
146. Asabe S, Wieland SF, Chattopadhyay PK. et al. The Size of the Viral Inoculum Contributes to the Outcome of Hepatitis B Virus Infection. J Virol. 2009;83:9652-9662
147. Itoh Y, Kuroda S, Miyazaki T. et al. Identification of Polymerized-Albumin Receptor Domain in the Pre-S2 Region of Hepatitis B Virus Surface Antigen M Protein. J Biotechnol. 1992;23:71-82
148. Carman WFF, Zanetti ARR, Karayiannis P. et al. Vaccine-Induced Escape Mutant of Hepatitis B Virus. Lancet. 1990;336:325-329
149. Waters JA, Kennedy M, Voet P. et al. Loss of the common “A” determinant of Hepatitis B Surface Antigen by a Vaccine-Induced Escape Mutant. J Clin Invest. 1992;90:2543-2547
150. Seitz S, Iancu C, Volz T. et al. A Slow Maturation Process Renders Hepatitis B Virus Infectious. Cell Host Microbe. 2016;20:25-35
151. Yamada T, Iwasaki Y, Tada H. et al. Nanoparticles for the Delivery of Genes and Drugs to Human Hepatocytes. Nat Biotechnol. 2003;21:885-890
152. Matsuura Y, Yagi H, Matsuda S. et al. Human Liver-Specific Nanocarrier in a Novel Mouse Xenograft Model Bearing Noncancerous Human Liver Tissue. Eur Surg Res. 2011;46:65-72
153. Jung J, Matsuzaki T, Tatematsu K. et al. Bio-Nanocapsule Conjugated with Liposomes for in Vivo Pinpoint Delivery of Various Materials. J Control Release. 2008;126:255-264
154. Kasuya T, Jung J, Kinoshita R. et al. Bio-Nanocapsule-liposome Conjugates for in Vivo Pinpoint Drug and Gene Delivery. Methods in Enzymology. Elsevier Inc. 2009;464:147-166
155. Liu Q, Jung J, Somiya M. et al. Virosomes of Hepatitis B Virus Envelope L Proteins Containing Doxorubicin: Synergistic Enhancement of Human Liver-Specific Antitumor Growth Activity by Radiotherapy. Int J Nanomedicine. 2015;10:4159-4172
156. Somiya M, Kuroda S. Development of a Virus-Mimicking Nanocarrier for Drug Delivery Systems: The Bio-Nanocapsule. Adv Drug Deliv Rev. 2015;95:77-89
157. Rodriguez-Crespo I, Gomez-Gutierrez J, Nieto M. et al. Prediction of a Putative Fusion Peptide in the S Protein of Hepatitis B Virus. J Gen Virol. 1994;75:637-639
158. Oess S, Hildt E. Novel Cell Permeable Motif Derived from the PreS2-Domain of Hepatitis-B Virus Surface Antigens. Gene Ther. 2000;7:750-758
159. Delgado CL, Núñez E, Yélamos B. et al. Study of the Putative Fusion Regions of the preS Domain of Hepatitis B Virus. Biochim Biophys Acta - Biomembr. 2015;1848:895-906
160. Liu Q, Somiya M, Shimada N. et al. Mutational Analysis of Hepatitis B Virus Pre-S1 (9-24) Fusogenic Peptide. Biochem Biophys Res Commun. 2016;474:406-412
Author contact
![]() Corresponding author: Prof. Shun'ichi Kuroda, Ph.D., Department of Biomolecular Science and Reaction, The Institute of Scientific and Industrial Research, Osaka University Mihogaoka 8-1, Ibaraki, Osaka 567-0047, Japan Tel: +81-6-6879-8460 FAX: +81-6-6879-8464 E-mail: skurodaosaka-u.ac.jp
Corresponding author: Prof. Shun'ichi Kuroda, Ph.D., Department of Biomolecular Science and Reaction, The Institute of Scientific and Industrial Research, Osaka University Mihogaoka 8-1, Ibaraki, Osaka 567-0047, Japan Tel: +81-6-6879-8460 FAX: +81-6-6879-8464 E-mail: skurodaosaka-u.ac.jp