- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
NMs from blood circulation to...
The EPR effect of NMs
Receptor-mediated targeting of...
Estimation of the relative...
Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(4):346-357. doi:10.7150/ntno.19380 This issue Cite
Review
Be Active or Not: the Relative Contribution of Active and Passive Tumor Targeting of Nanomaterials
Rui Li1, Ke Zheng2, Cai Yuan2, Zhuo Chen3, Mingdong Huang2, 3 ![]()
1. Key Laboratory of Animal Immunology of the Ministry of Agriculture, Henan Provincial Key Laboratory of Animal Immunology, Henan Academy of Agricultural Sciences, Zhengzhou, Henan, 450002, China;
2. Fuzhou University, Fuzhou, Fujian, 350116, China;
3. Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian, 350002, China.
Received 2017-1-31; Accepted 2017-6-21; Published 2017-7-11
Abstract

Malignant tumor (cancer) remains as one of the deadliest diseases throughout the world, despite its overall mortality drops. Nanomaterials (NMs) have been widely studied as diagnostic and/or therapeutic agents for tumors. A feature of NMs, compared to small molecules, is that NMs can be concentrated passively in tumors through enhanced permeability and retention (EPR) effect. In the meantime, NMs can be engineered to target toward tumor specific markers in an active manner, e.g., receptor-mediated targeting. The relative contribution of the EPR effect and the receptor-mediated targeting to NM accumulation in tumor tissues has not been clearly defined yet. Here, we tackle this fundamental issue by reviewing previous studies. First, we summarize the current knowledge on these two tumor targeting strategies of NMs, and on how NMs arrive to tumors from blood circulation. We then demonstrate that contribution of the active and passive effects to total accumulation of NMs in tumors varies with time. Over time, the receptor-mediated targeting contributes more than the EPR effect with a ratio of 3 in the case of urokinase-type plasminogen activator receptor (uPAR)-mediated targeting and human serum albumin (HSA)-mediated EPR effect. Therefore, this review highlights the dynamics of active and passive targeting of NMs on their accumulation at tumor sites, and is valuable for future design of NMs in cancer diagnosis and treatment.
Keywords: nanomaterial, nanoparticle, enhanced permeability and retention effect, receptor-mediated tumor targeting, urokinase-type plasminogen activator receptor, amino-terminal fragment.
Introduction
The past decades have witnessed continuous advances in tumor diagnosis and therapies; however, malignant tumor (cancer) remains as one of the deadliest diseases throughout the world. More than 1.6 million new cancer cases and over 0.6 million cancer deaths are estimated to occur in the United States alone in 2017, despite the overall mortality drops [1].
Nanomaterials (NMs), including nanoparticles (NPs), micelles, dendrimers and liposomes, have emerged as a novel class of diagnostic probes and/or therapeutic drugs for tumors [2-4]. The nano-sized dimension renders NMs with unique physicochemical properties, such as optical properties and high ratio of surface area to volume. In addition, NMs can be engineered to be responsive to environmental conditions, such as pH value, redox potential or temperature, for controlled release of imaging agents or drugs. Additional properties can be further engineered to NMs, such as biocompatibility, bioavailability, and tumor selectivity [5-8].
Doxil® and Abraxane® represent two successful clinical applications of NMs in cancer nanomedicine. Doxil® is a polyethylene glycol coated (pegylated) liposome-encapsulated doxorubicin (DOX), and was approved by the US Food and Drug Administration (FDA) for treatment of AIDS-related Kaposi's sarcoma, ovarian cancer and multiple myeloma [9]. DOX is a highly potent drug used in tumor chemotherapy, but has poor tumor specificity and is toxic to normal tissues, especially cardiomyocytes [10, 11]. Compared to free DOX, Doxil® is concentrated preferentially in tumors and has a better therapeutic index [12, 13]. Abraxane® is albumin-bound paclitaxel NP and is approved by the FDA for the treatment of breast cancer [14], non-small-cell lung cancer [15] and other solid tumors [16, 17]. The formulation of paclitaxel with albumin into NPs renders water solubility of hydrophobic paclitaxel and avoids the use of the solubilizing agent Cremophor which often causes hypersensitivity reactions [18]. Abraxane® is specially accumulated at tumor sites, which reduces its cytotoxicity to normal tissues and thus increases its maximum tolerated dose. Some other NM formulations, including dendrimers [19, 20], quantum dots (QDs) [21-24], metal NPs (e.g., magnetic iron oxide [25] and gold [26]), are under study for cancer nanomedicine [27].
NMs can target to tumors either in a passive manner through the enhanced permeability and retention (EPR) effect or in an active manner by receptor-mediated targeting [28-31]. The relative contribution of the EPR effect and receptor-mediated targeting to NM accumulation in tumor is not fully defined. In this review, we first describe how NMs arrive at tumor sites from blood circulation in vivo. Next we briefly describe the EPR effect and receptor-mediated targeting. Finally, we try to estimate the relative contribution of the EPR effect and receptor-mediated targeting of NMs by summarizing the results reported in the literature.
NMs from blood circulation to tumors
For in vivo application, NMs are typically delivered or redistributed to tumors through blood circulation in vascular systems. In normal tissues, molecular exchange across vasculature takes place primarily in capillaries, which consist of a layer of endothelial cells and occasional connective tissue (Figure 1) [32, 33]. Molecules smaller than 3 nm, such as water, gases, salts, sugars and certain metabolites pass capillary endothelial cells freely, largely by diffusion through the space between adjacent capillary endothelium (intercellular cleft, Figure 1) or transcytosis [33, 34]. Molecules larger than 3 nm cannot pass through endothelium freely and only a small amount of macromolecules, such as albumin, immunoglobulins (Igs) and other plasma proteins, are found to extravasate from circulation into normal tissues [32, 34].
Schematic diagram of the blood circulatory system. There are three major types of blood vessels: arteries, veins and capillaries. Capillaries are consisted of a layer of endothelial cells, surrounded by basement membranes.
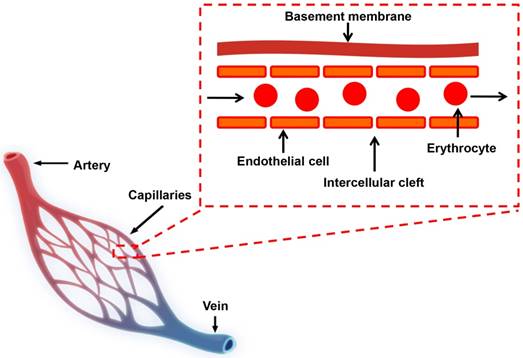
Under some circumstances like inflammation, large molecules can exit vasculature in quantity. This occurs primarily in post-capillary venules [35, 36]. The transcellular passage of large molecules may be through enlarged intercellular cleft induced by vascular permeabilizing factors [34]. Another potential mechanism of extravasation is through the vesiculo-vacuolar organelle (VVO). The venular endothelial cells are cuboidal and characterized by clusters of interconnected vesicles and vacuoles in their cytoplasm, distinctive from capillary endothelial cells. These intracellular vesicles and vacuoles together form VVOs [37-39]. The VVOs are linked to the plasma membrane by stomata that are normally closed by thin diaphragms [40]. When exposed to vascular permeabilizing factors, stromal diaphragms are pulled apart mechanically and VVOs are open, allowing transcellular passage of large molecules [41]. The vascular permeabilizing factors include vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) [42-46], histamine [35, 36], serotonin [35] and platelet activating factor. A single exposure to any of these permeability factors results in a rapid (~20-30 min) hyperpermeability.
Solid tumors stimulate the formation of new blood vessels (neovasculature) in order to absorb excessive nutrients and proliferate quickly [47]. Extensive angiogenesis and high vascular density are hallmarks of solid tumors [48]. The neovasculature is quite different from normal vasculature both in form and in architecture. The vascular basement membranes (VBMs) of neovasculature are easily degraded by various proteases (the matrix metalloproteases and the plasminogen activator) [49], allowing the detachment of pericytes from vascular endothelial cells. The endothelial cells, no longer restrained by VBMs or pericytes, become thin as their lumens expand in response to intravascular pressure [50]. In addition, the endothelial cells are poorly aligned with wide fenestrations; both smooth muscle layer and innervation are malformed or even absent, and functional receptors for some modulators, e.g., angiotensin II (AT-II), are missing [51]. These characteristics ultimately lead to a highly leaky vasculature. Large molecules thus can extravasate out of these blood vessels and into tumors as a result of transvascular osmotic pressure [39]. This increased vascular permeability is chronic and can be utilized to deliver NMs to solid tumors. In the meantime, it is worth to mention that this tumor related permeability is dynamic instead of static [52].
Once reaching tumor sites, NMs need to traverse into intracellular space (endocytosis or cellular trafficking) to take effect. This is a critical event affecting the efficacy and specificity of NMs, and has been widely studied or reviewed [53-56].
The EPR effect of NMs
A term of “enhanced permeability and retention (EPR) effect” was firstly proposed by Matsumura and Maeda in 1986 to describe the preferential accumulation of macromolecules in tumor, which has leaky capillaries, and at the same time, poor lymphatic drainage [57]. Size or molecular weight of the macromolecule is a key parameter determining the EPR effect in solid tumors (Figure 2). Typically, molecules with sizes ranging from 10 to 200 nm or 40 to 800 kilodaltons (kDa) in mass exhibit a strong EPR effect [58-60]. Such macromolecules include plasma proteins and NMs [61, 62]. Small molecules tend to diffuse freely in and out of tumor blood vessels because of their small sizes, and thus do not accumulate in tumors as much as macromolecules do over time (Figure 2) [63]. Besides the size, other properties, such as shape and surface chemistry, also affect the EPR effect of NMs, which are discussed in recent reviews [31, 64] and will not be elaborated here.
Schematic illustration of the enhanced permeability and retention (EPR) effect in tumors. In tumor tissues (lower panels), endothelial cells are poorly aligned with wide fenestrations, and there is a lack of lymphatic clearance and a smooth muscle layer compared to normal tissues (top panels). Therefore, macromolecules (10 to 200 nm or 40 to 800 kDa) tend to accumulate in tumor tissues much more than in normal tissues. In contrast, small molecules diffuse freely in and out of blood vessels in both normal and tumor tissues due to their small sizes, leading to their low concentrations over time.
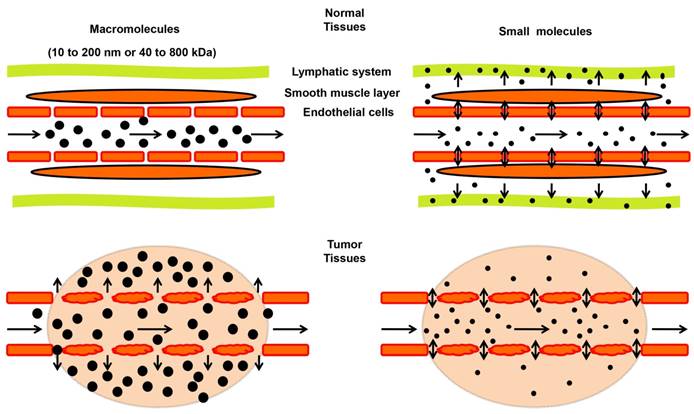
The EPR effect can be modulated by a number of in vivo factors. EPR augmenting factors include (1) vasoconstrictors to raise the systemic blood pressure [65], e.g., AT-II [66]; (2) free radicals that affect integrity of vascular endothelium, e.g., peroxynitrite [67]; (3) nitric oxide-releasing agents, e.g., nitroglycerin [68]; (4) vascular permeability promoters: bradykinin/kinin [69], prostaglandins, VEGF/VPF and other inflammatory cytokines [70]. These stimulators result in an enhanced vascular permeability and extravasation of macromolecules, and thus increase the EPR effect.
There are a number of studies on tumor-targeted NMs mediated by the EPR effect, which were summarized in other reviews [31, 71]. Therefore, only a few representative examples are included in this review. In early 1980s, Maeda and his colleagues prepared a polymer (poly(styrene-comaleic acid)/half-n-butyl ester, SMA) conjugated with a derivative of a DNA-damaging agent (neocarzinostatin, NCS) [72], designated as SMANCS [73]. SMANCS is small in mass (16 kDa) but binds to albumin (67 kDa) in blood circulation [74], leading to an apparent molecular mass of about 83 kDa. The conjugates achieved not only high contrast in tumor imaging when combined with an imaging agent (Lipiodol®), but also a strong anti-tumor therapeutic response [57]. The clinical use of SMANCS was approved in 1993 by the Japanese Government for hepatoma treatment [75]. Doxil® and Abraxane® with ~100 nm in size [76, 77], also have their tumor targeting attributed to the EPR effect.
A few drawbacks, however, exist for the EPR effect-mediated tumor targeting of NMs. The delivery of NMs to tumors through the EPR effect was reported to be inefficient and provides only 20-30% increases in delivery compared with normal organs [64]. This inefficiency is probably because of tumor heterogeneity. Solid tumors are highly dynamic and complex with heterogeneity on size, genomic makeup, vasculature, interstitial pressures and necrotic cores [78-81]. In addition, the EPR effect has modest specificity, because high vascular permeability also occurs under other pathological conditions, e.g., inflammation [28].
Receptor-mediated targeting of NMs
Tumor cells express a profile of surface receptors different from normal cells (Figure 3) [48]. Receptor-mediated targeting of NMs to tumors takes advantage of these specific receptors, which potentially has high safety margin by reducing damage to normal tissues [28, 82-86]. Commonly used tumor surface receptors include epidermal growth factor receptor (EGFR) [87], human epidermal growth factor receptor 2 (HER2) [87], transferrin receptor [88, 89], folate receptor [90, 91], integrins [92] and urokinase-type plasminogen activator (uPA) receptor (uPAR) [93-96]. A complete list of targeted receptors can be found in recent reviews [97, 98].
Schematic illustration of receptor-mediated targeting in tumors. Tumor cells express a profile of surface receptors different from normal cells.
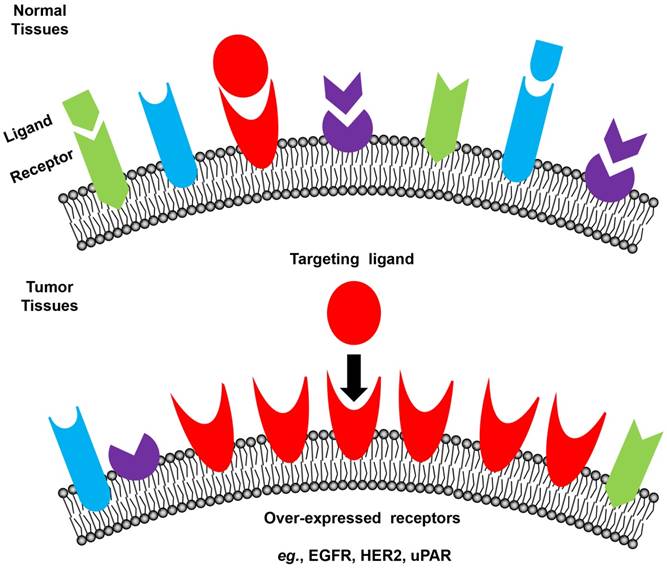
For example, EGFR, a member of the ErbB family of receptor tyrosine kinases, is a widely targeted receptor for clinical applications [99-102]. Aberrant EGFR over-expression was reported in tumors, including lung cancer [103, 104], pancreatic cancer [105], and glioblastoma [106]. Monoclonal anti-EGFR antibodies (mAbs) cetuximab [107-110] and panitumumab [111, 112] are both approved by the FDA, originally for metastatic colorectal cancer. A list of other mAbs (zalutumumab, nimotuzumab and matuzumab) is in clinical development [113, 114]. HER2, another member of the ErbB family, is also successfully explored for tumor targeting [115]. Trastuzumab is effective toward breast cancers where HER2 is over-expressed [116]. Pertuzumab was approved by the FDA for use in combination with trastuzumab in 2012 [117].
uPAR is expressed in a low level on the surfaces of quiescent cells, but is greatly over-expressed on the surfaces of a wide range of invasive tumor cells, and is believed to play a critical role in tumor cell migration, adhesion and tissue remodeling [118-121]. In fact, uPAR has been studied as a promising candidate receptor in tumor targeting therapy [122-127] and/or imaging [128-133]. A peptidyl antagonist of uPAR was successfully used for nuclear imaging of primary tumors and lymph node metastases in 10 patients, and its uptakes were found to correlate with high uPAR expression in excised tumor tissues [134].
Receptor-mediated NMs have been widely demonstrated for their tumor targeting efficacies in vitro. The representative examples are listed in Table 1. The gold NPs with the average size of 35 nm conjugated with an anti-EGFR antibody were shown to specifically bind to cancer cells with 6-fold greater affinity than to the noncancerous cells [135]. The cytotoxicity of a drug formulated in NPs decorated with trastuzumab targeting at HER2 (around 300 nm) was found to be 12.7-fold higher than that of the bare ones without the mAb [136]. The cytotoxic paclitaxel embedded in transferrin-conjugated NPs (about 220 nm) targeting at transferrin receptor was shown a 3-fold higher uptake by human prostate cancer cells than the unconjugated ones [137]. An NM construct was prepared by conjugating magnetic iron oxide NPs with the amino-terminal fragment (ATF) of uPA, which is a highly potent (~0.2 nM dissociation constant) peptide targeting to uPAR [138]. As a result, these conjugated NPs (66 nm) exhibited 7-fold higher accumulation in pancreatic cancer cells than unconjugated ones [139].
Selected examples of receptor-mediated targeting for NMs.
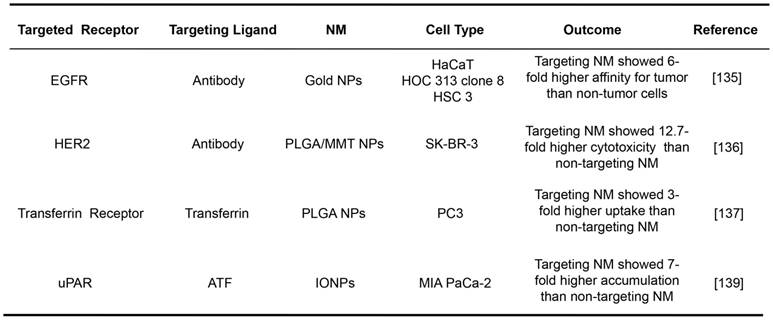
HaCaT: nonmalignant epithelial cell line; HOC 313 clone 8/HSC 3: malignant oral epithelial cell lines; PLGA/MMT NPs: poly(D,L-lactide-co-glycolide)/montmorillo- nite nanoparticles; SK-BR-3: breast cancer cell line; PC3: prostate cancer cell line; IONPs: magnetic iron oxide nanoparticles; MIA PaCa-2: pancreatic cancer cell line.
Two parameters are critical for the success of receptor-mediated targeting strategy. One is the abundance of tumor surface receptors compared to that in normal tissues. Such abundance typically undergoes active regulation at transcriptional/translational levels or through receptor endocytosis. Genetic mutations may alter receptor structures and/or functions, and thus lower or disrupt the efficacies of specific diagnostic agents or therapeutic drugs, leading to drug resistance [84, 140-142]. The other parameter is the targeting agents for the receptors. The potency, specificity, molecular display, pharmacodynamics, pharmacokinetics and safety profiles should be taken into account for the choice of targeting agents. The common targeting agents include small molecules, aptamers, peptides, proteins, and antibodies, which can be roughly classified into two categories based on size/molecular weight with small organic molecules and mAbs as representative member (Table 2). The molecular mass of small organic molecules tends to be below 500 Daltons as summarized by Lipinski's rule of five [143, 144]. Though generally used in clinical practice, small molecular agents bear one inherent concern on their target specificity. mAbs have high molecular mass, e.g., ~160 kDa for IgG, and belong to NMs in term of their sizes (a hydrodynamic radius of 7-12 nm for an IgG molecule). They tend to have strong potencies and high specificities to their targets, but have potential antigenicity and poor oral bioavailability. The conventional administration route for mAbs is through intravenous or subcutaneous delivery [145]. The importance of these two parameters for a successful target has been clarified in a recent review [146].
Examples of targeting agents for tumor receptor-mediated NMs.

The corresponding receptor targets are listed in the parentheses.
Estimation of the relative contribution between the EPR effect and receptor-mediated targeting of NMs
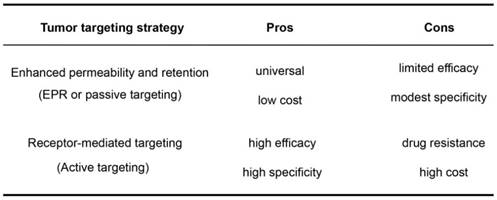
As both tumor-targeting strategies have their pros and cons (Table 3), a combination of these two strategies may add value. Supplementing the EPR effect of NMs with receptor-mediated targeting may reduce the effect of tumor heterogeneity as well as enhance selectivity and efficacy of NMs against solid tumors. Despite the prevalent development of such tumor-targeting NMs, a question remains: what are the relative contribution between the EPR effect and receptor-mediated effects to tumor targeting in vivo?
The pros and cons of the EPR effect (passive targeting) and receptor-mediated targeting (active targeting) for NM accumulation in tumors.
In an attempt to resolve this issue, our group constructed a novel tumor targeting NM: a recombinant protein of human serum albumin (HSA, 67 kDa, 6.5 nm) fused with a tumor targeting agent (ATF, 14 kDa) at the N-terminus of HSA (labeled as ATF-HSA, 84 kDa, 7.5 nm, Figure 4) [147]. HSA is previously demonstrated to accumulate in tumor tissues mainly through the EPR effect [57, 148, 149]. Although it was proposed that HSA might be internalized into cells through a protein named SPARC (secreted protein, acidic and rich in cysteine) [150], the in vivo significance of SPARC is not demonstrated in a convincible way, considering that the large quantity of endogenous HSA in blood circulation may saturate most of HSA binding proteins. HSA is also recognized as nano-sized drug carrier capable of embedding small molecule drugs [151-153]. ATF-HSA thus exhibits dual targeting modes: its ATF peptide targets potently at uPAR on tumor surface and its large molecular size targets at tumor through the EPR effect. For comparison, we used HSA as a control and assumed HSA has the EPR effect comparable to ATF-HSA due to their similar molecular sizes (~7 nm).
Schematic illustration of the estimation of the relative contribution between the EPR effect and receptor-mediated targeting for NMs. A novel tumor targeting NM was constructed of a recombinant protein of human serum albumin (HSA) fused with a tumor receptor targeting agent (ATF) at the N-terminus of HSA (labeled as ATF-HSA). ATF-HSA exhibits dual targeting modes: Its ATF peptide targets at uPAR over-expressed on tumor surfaces and HSA targets at tumor through the EPR effect. HSA was used as a control, which has only the EPR effect but no receptor targeting effect.
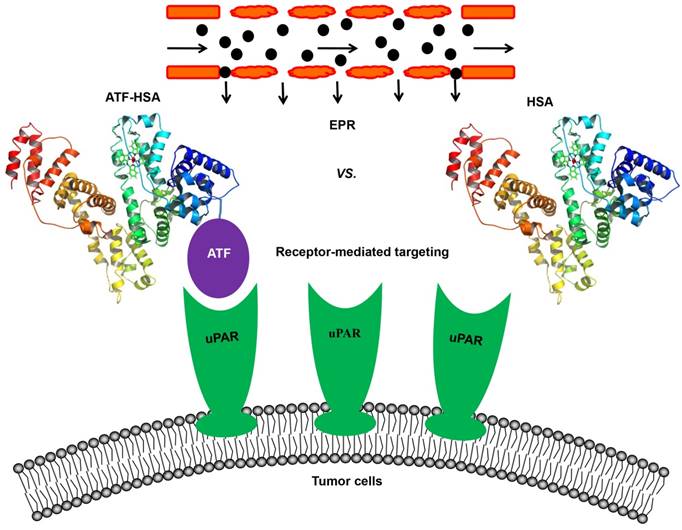
To visualize and quantify the accumulation of the targeting NM in tumors, we loaded a near-infrared fluorescent imaging probe (mono-substituted β-carboxyl phthalocyanine zinc, abbreviated as CPZ [154]) into either HSA or ATF-HSA to form nano-sized complexes (Figure 4). CPZ has a maximal absorption coefficient at 680 nm, which is optimal for deep tissue penetration (up to 10 mm), and for three-dimensional non-invasive and quantitative optical imaging using fluorescent molecular tomography method. It should be emphasized that the loading of the CPZ is not through covalent linkage, and the embedding of the CPZ inside HSA or ATF-HSA does not change the tumor targeting properties or sizes of macromolecules [147]. The CPZ was bound quite tightly inside the proteins and was stable in saline and plasma without the bleaching of its blue color or the degradation of protein [147].
We then measured the fluorescent signals of HSA or ATF-HSA on Hepatoma-22 (H22)-bearing Kunming mice, which express a high level of murine uPAR (Figure 5A) [155]. As seen in Figure 5B, at 6 h and 12 h post-injection of 0.05 mg CPZ/kg of mouse body weight via caudal vein, both HSA and ATF-HSA accumulated at the tumor sites with ~3-fold higher concentrations compared to the non-tumor sites. At these two time points, both ATF-HSA and HSA had almost the same amount at tumor sites. Such tumor accumulation is most likely due to the EPR effect. At 24 h post-injection, ATF-HSA showed a higher amount of tumor accumulation than HSA (~2 folds), with HSA accumulation in tumors remained at about 3-fold comparing to non-tumor accumulation. This difference is likely due to uPAR-mediated targeting, which takes effect following the EPR effect in vivo. At 96 h post-injection, this tumor retention difference was further enlarged and ATF-HSA showed almost 3 times more tumor accumulation compared to HSA, which demonstrated the tumor targeting contribution of the receptor-mediated effect and the EPR effect were 75% and 25%, respectively.
The relative contribution of the EPR effect and receptor-mediated targeting effect to tumor accumulation of NMs. (A) Specific accumulation of both HSA (top panels) and ATF-HSA (bottom panels) in tumors are shown in the front view of the representative mice and three-dimensional view of tumors 48 h post-injection to H22 tumor-bearing Kunming mice. (B) Dynamics of tumor targeting of HSA (blank bars) and ATF-HSA (shaded bars) on H22 tumor-bearing mice. Average concentrations of fluorescent probe (CPZ) were quantitated by non-invasive fluorescent molecular tomography at the tumor and non-tumor sites at indicated time points after injection to the tumor-bearing mice. Both HSA and ATF-HSA accumulated rapidly at the tumors sites, as seen from the data at 6 h and 12 h, with signals of 3 folds compared to the non-tumor sites. After 24 h injection, ATF-HSA showed higher tumor retention than HSA (about 2 times). At 96 h post-injection, ATF-HSA showed almost 4 times tumor accumulation compared to HSA. The data were averaged from 3 mice in each group at each time point; bars represent standard error of the mean (SEM). *Significantly higher CPZ tumor accumulation for ATF-HSA compared to that of HSA, p < 0.05.
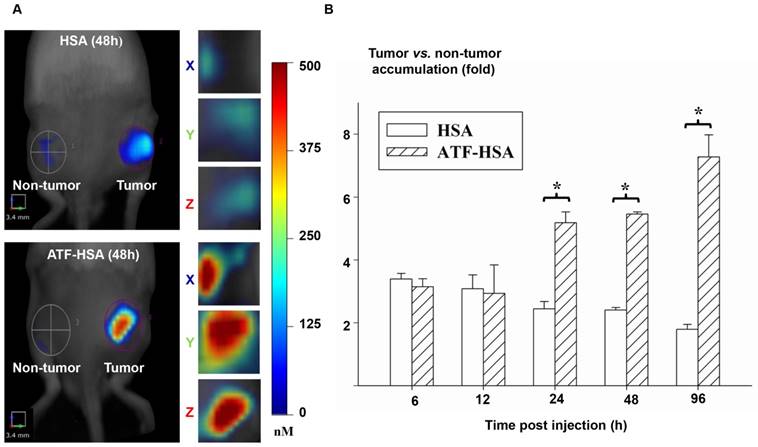
The main conclusions we extracted from these results (Figure 5) are: 1) for once infusion, the tumor accumulation of NMs through the passive targeting (the EPR effect) reaches a plateau (at least 3-fold compared to non-tumor accumulation) at around 24 h, but gradually reduces after 48 h; 2) the tumor accumulation due to the active targeting becomes prominent at 24 h post-injection, and increases continuously up to 96 h; 3) the relative ratio of the active to passive targeting effect depends on time after the NM injection, which is about 1 at 24 h and 3 at 96 h.
To further validate these results, we used murine ATF (mATF) as targeting agent which binds more tightly to surface uPAR of xenografted murine tumors [156]. The imaging probe loaded mATF-HSA and ATF-HSA have nearly identical shapes, charges and molecular masses (82 kDa and 84 kDa, respectively), and are therefore likely to have the identical EPR effect. We observed that the CPZ average concentration of mATF-HSA group in tumor sites was significantly higher (at least 2 times) than that of the ATF-HSA group 6 h post-injection (especially at 12 h and 24 h), demonstrating a higher contribution of receptor-mediated targeting compared to the EPR effect (6-fold, figure not shown) [156].
Our estimation of this relative contribution certainly has some limitations. We used a transplanted murine tumor model and the results need to be validated in other tumor models, e.g., in situ nascent tumors. It will be useful to include more time points in active/passive targeting measurements to validate our estimation. Moreover, NMs with different compositions or radii, and additional readout methods, e.g., positron emission tomography or magnetic resonance imaging, need to be explored. The tumor targeting NMs used in our current studies are proteins, which are soft, and this estimation may be different from that for rigid NMs.
To our knowledge, there are various controversial in vivo data on estimation of this relative contribution in literature. In one study, NPs of super-paramagnetic iron oxide (SPIO) were functionalized with tripeptide arginine-glycine-aspartic acid (RGD) targeting to integrin αvβ3 with a size of about 240 nm [157]. Their bio-distributions were detected in mice inoculated subcutaneously with CT26 colon carcinoma cells using electron spin resonance spectroscopy. The mice injected with RGD-functionalized SPIO NPs showed a 2.5-fold higher signal in tumor tissues compared to the mice treated with bare NPs 1 h and 4 h post-injection [157]. This study thus demonstrated the receptor-mediated tumor targeting contributed 2.5-fold more than the EPR effect, which is consistent with our results. In another study, active accumulation of transferrin-coated gold nanoparticles (60 nm) was found to be 5 times faster and approximately 2-fold higher relative to their passive poly(ethylene glycol)-coated counterparts in MDAMB-435 orthotopic tumor xenografts [158]. However, in one study using cyclic RGD-conjugated gold nanorods (about 80 nm), the total tumor accumulation of these so-called targeting NMs was found to be only marginally improved compared to non-targeting ones [159]. Another study using folate as targeting agent showed that the targeting liposomes (in the range of 70 to 90 nm) did not enhance the overall liposome deposition in tumors [160]. A comprehensive analysis of NP delivery to tumors based on studies published between 2005 and 2015 showed that the NMs using active targeting had a slight higher efficiency than those relied on the passive targeting (0.9% versus 0.6%) [161]. Taken together, further studies are definitely needed to address this important issue of active vs. passive targeting, and to gain further mechanism insights.
Perspectives
NMs are widely studied as diagnostic and/or therapeutic agents targeting tumors in a passive manner through the EPR effect, in an active strategy by receptor-mediated targeting, or with a strategy combining these two. However, cancer nanomedicine is under debate recently [162]. It appears that tumor targeting NMs need to be re-examined [163, 164].
Here, our studies showed the relative contribution of active and passive targeting varied with time, and active targeting appeared contributing more than passive targeting over time. The nature of targeting agents and tumor receptors is of course a critical variable determining the ratio between these two targeting effects [159]. The observed dynamics of active and passive targeting also directly affects the efficacies of NMs, which is an important factor to be considered in the design of NMs. As a consequence, further studies on the dynamics, and the underlying mechanisms, including the factors affecting the dynamics, are highly advocated [165].
Abbreviations
ATF: amino-terminal fragment of uPA; CPZ: β-carboxy phthalocyanine zinc; EPR: enhanced permeability and retention; HSA: human serum albumin; NM: nanomaterial; NP: nanoparticle; uPA: urokinase-type plasminogen activator; uPAR: uPA receptor.
Acknowledgements
This work was supported by grants from Natural Science Foundation of China (31370737, U1405229, 31602036), Henan Province (the Basic and Advanced Technology Research Program 162300410252) and Henan Academy of Agricultural Sciences ([2017]76-22).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Emerich DF. Nanomedicine-prospective therapeutic and diagnostic applications. Expert Opin Biol Ther. 2005;5:1-5
3. Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211-7
4. Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology applications in cancer. Annu Rev Biomed Eng. 2007;9:257-88
5. LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21:1184-91
6. Minchin R. Nanomedicine: sizing up targets with nanoparticles. Nat Nanotechnol. 2008;3:12-3
7. Conde J, de la Fuente JM, Baptista PV. Nanomaterials for reversion of multidrug resistance in cancer: a new hope for an old idea? Front Pharmacol. 2013;4:134
8. Jiang S, Win KY, Liu S, Teng CP, Zheng Y, Han MY. Surface-functionalized nanoparticles for biosensing and imaging-guided therapeutics. Nanoscale. 2013;5:3127-48
9. Barenholz Y. Doxil(R)-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-34
10. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R. et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun. 2013;4:1908
11. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157-70
12. Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O. et al. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11:1029-33
13. Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19:424-36
14. Miele E, Spinelli GP, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99-105
15. Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P. et al. Abraxane, a novel Cremophor-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann Oncol. 2006;17:1263-8
16. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7:1041-53
17. Stinchcombe TE, Socinski MA, Walko CM, O'Neil BH, Collichio FA, Ivanova A. et al. Phase I and pharmacokinetic trial of carboplatin and albumin-bound paclitaxel, ABI-007 (Abraxane) on three treatment schedules in patients with solid tumors. Cancer Chemother Pharmacol. 2007;60:759-66
18. Damascelli B, Cantu G, Mattavelli F, Tamplenizza P, Bidoli P, Leo E. et al. Intraarterial chemotherapy with polyoxyethylated castor oil free paclitaxel, incorporated in albumin nanoparticles (ABI-007): Phase I study of patients with squamous cell carcinoma of the head and neck and anal canal: preliminary evidence of clinical activity. Cancer. 2001;92:2592-602
19. Nanjwade BK, Bechra HM, Derkar GK, Manvi FV, Nanjwade VK. Dendrimers: emerging polymers for drug-delivery systems. Eur J Pharm Sci. 2009;38:185-96
20. Astruc D, Boisselier E, Ornelas C. Dendrimers designed for functions: from physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem Rev. 2010;110:1857-959
21. Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Noninvasive imaging of quantum dots in mice. Bioconjug Chem. 2004;15:79-86
22. Howarth M, Liu W, Puthenveetil S, Zheng Y, Marshall LF, Schmidt MM. et al. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nat Methods. 2008;5:397-9
23. Wang FB, Rong Y, Fang M, Yuan JP, Peng CW, Liu SP. et al. Recognition and capture of metastatic hepatocellular carcinoma cells using aptamer-conjugated quantum dots and magnetic particles. Biomaterials. 2013;34:3816-27
24. Das M, Duan W, Sahoo SK. Multifunctional nanoparticle-EpCAM aptamer bioconjugates: a paradigm for targeted drug delivery and imaging in cancer therapy. Nanomedicine. 2015;11:379-89
25. Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L. et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108:2064-110
26. Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38:1759-82
27. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
28. Arias JL. Drug targeting strategies in cancer treatment: an overview. Mini Rev Med Chem. 2011;11:1-17
29. Barreto JA, O'Malley W, Kubeil M, Graham B, Stephan H, Spiccia L. Nanomaterials: applications in cancer imaging and therapy. Adv Mater. 2011;23:H18-40
30. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2-25
31. Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116:5338-431
32. Rippe B, Haraldsson B. Transport of macromolecules across microvascular walls: the two-pore theory. Physiol Rev. 1994;74:163-219
33. Curry FR. Microvascular solute and water transport. Microcirculation. 2005;12:17-31
34. Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109-19
35. Majno G, Palade GE, Schoefl GI. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: a topographic study. J Biophys Biochem Cytol. 1961;11:607-26
36. Majno G, Shea SM, Leventhal M. Endothelial contraction induced by histamine-type mediators: an electron microscopic study. J Cell Biol. 1969;42:647-72
37. Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med. 1996;183:1981-6
38. Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 1999;6:23-44
39. Feng D, Nagy JA, Dvorak AM, Dvorak HF. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res. 2000;59:24-37
40. Feng D, Nagy JA, Hipp J, Pyne K, Dvorak HF, Dvorak AM. Reinterpretation of endothelial cell gaps induced by vasoactive mediators in guinea-pig, mouse and rat: many are transcellular pores. J Physiol. 1997;504( Pt 3):747-61
41. Stan RV. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J Cell Mol Med. 2007;11:621-43
42. Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986;46:5629-32
43. Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S. et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99-115
44. Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2002;39:225-37
45. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368-80
46. Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002;29:10-4
47. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-64
48. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
49. Chang SH, Kanasaki K, Gocheva V, Blum G, Harper J, Moses MA. et al. VEGF-A induces angiogenesis by perturbing the cathepsin-cysteine protease inhibitor balance in venules, causing basement membrane degradation and mother vessel formation. Cancer Res. 2009;69:4537-44
50. Nagy JA, Dvorak HF. Heterogeneity of the tumor vasculature: the need for new tumor blood vessel type-specific targets. Clin Exp Metastasis. 2012;29:657-62
51. Kohn S, Nagy JA, Dvorak HF, Dvorak AM. Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels. Lab Invest. 1992;67:596-607
52. Matsumoto Y, Nichols JW, Toh K, Nomoto T, Cabral H, Miura Y. et al. Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat Nanotechnol. 2016;11:533-8
53. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB. et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732-40
54. Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME. et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613-8
55. Tekle C, Deurs B, Sandvig K, Iversen TG. Cellular trafficking of quantum dot-ligand bioconjugates and their induction of changes in normal routing of unconjugated ligands. Nano Lett. 2008;8:1858-65
56. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145:182-95
57. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
58. Babson AL, Winnick T. Protein transfer in tumor-bearing rats. Cancer Res. 1954;14:606-11
59. Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K. et al. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur J Cancer. 1995;31A:766-70
60. Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T. et al. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. 1998;89:307-14
61. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25-37
62. Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138-44
63. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818-22
64. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81-9
65. Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol. 2009;39:756-66
66. Hori K, Suzuki M, Tanda S, Saito S, Shinozaki M, Zhang QH. Fluctuations in tumor blood flow under normotension and the effect of angiotensin II-induced hypertension. Jpn J Cancer Res. 1991;82:1309-16
67. Wu J, Akaike T, Hayashida K, Okamoto T, Okuyama A, Maeda H. Enhanced vascular permeability in solid tumor involving peroxynitrite and matrix metalloproteinases. Jpn J Cancer Res. 2001;92:439-51
68. Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100:2426-30
69. Wu J, Akaike T, Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998;58:159-65
70. Maeda H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:53-71
71. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
72. Maeda H, Takeshita J, Kanamaru R. A lipophilic derivative of neocarzinostatin. A polymer conjugation of an antitumor protein antibiotic. Int J Pept Protein Res. 1979;14:81-7
73. Maeda H, Ueda M, Morinaga T, Matsumoto T. Conjugation of poly(styrene-co-maleic acid) derivatives to the antitumor protein neocarzinostatin: pronounced improvements in pharmacological properties. J Med Chem. 1985;28:455-61
74. Peters T. Serum albumin. Adv Protein Chem. 1985;37:161-245
75. Maeda H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169-85
76. Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R. et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83-94
77. Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother. 2009;10:333-43
78. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883-92
79. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479-85
80. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
81. Crockford A, Jamal-Hanjani M, Hicks J, Swanton C. Implications of intratumour heterogeneity for treatment stratification. J Pathol. 2014;232:264-73
82. Jain RK. The next frontier of molecular medicine: delivery of therapeutics. Nat Med. 1998;4:655-7
83. Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615-26
84. Markman M. The promise and perils of 'targeted therapy' of advanced ovarian cancer. Oncology. 2008;74:1-6
85. Katzel JA, Fanucchi MP, Li Z. Recent advances of novel targeted therapy in non-small cell lung cancer. J Hematol Oncol. 2009;2:2
86. Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27:286-98
87. Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol. 2006;8:77-82
88. Sutherland R, Delia D, Schneider C, Newman R, Kemshead J, Greaves M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc Natl Acad Sci U S A. 1981;78:4515-9
89. Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144-58
90. Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54:675-93
91. Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94:2135-46
92. Hallahan D, Geng L, Qu S, Scarfone C, Giorgio T, Donnelly E. et al. Integrin-mediated targeting of drug delivery to irradiated tumor blood vessels. Cancer Cell. 2003;3:63-74
93. Li D, Liu S, Shan H, Conti P, Li Z. Urokinase plasminogen activator receptor (uPAR) targeted nuclear imaging and radionuclide therapy. Theranostics. 2013;3:507-15
94. O'Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics. 2013;3:496-506
95. Yang L, Sajja HK, Cao Z, Qian W, Bender L, Marcus AI. et al. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics. 2013;4:106-18
96. LeBeau AM, Sevillano N, King ML, Duriseti S, Murphy ST, Craik CS. et al. Imaging the urokinase plasminongen activator receptor in preclinical breast cancer models of acquired drug resistance. Theranostics. 2014;4:267-79
97. Chen H, Zhen Z, Todd T, Chu PK, Xie J. Nanoparticles for improving cancer diagnosis. Mater Sci Eng R Rep. 2013;74:35-69
98. Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG. et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19-34
99. Ennis BW, Lippman ME, Dickson RB. The EGF receptor system as a target for antitumor therapy. Cancer Invest. 1991;9:553-62
100. Kari C, Chan TO, Rocha de Quadros M, Rodeck U. Targeting the epidermal growth factor receptor in cancer: apoptosis takes center stage. Cancer Res. 2003;63:1-5
101. Scartozzi M, Pierantoni C, Berardi R, Antognoli S, Bearzi I, Cascinu S. Epidermal growth factor receptor: a promising therapeutic target for colorectal cancer. Anal Quant Cytol Histol. 2006;28:61-8
102. Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R. et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051-8
103. Ahmed SM, Salgia R. Epidermal growth factor receptor mutations and susceptibility to targeted therapy in lung cancer. Respirology. 2006;11:687-92
104. Prudkin L, Wistuba II. Epidermal growth factor receptor abnormalities in lung cancer. Pathogenetic and clinical implications. Ann Diagn Pathol. 2006;10:306-15
105. Cohenuram M, Saif MW. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present and the future. JOP. 2007;8:4-15
106. Nakamura JL. The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications. Expert Opin Ther Targets. 2007;11:463-72
107. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F. et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663-71
108. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG. et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563-72
109. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408-17
110. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M. et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466-75
111. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B. et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658-64
112. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ. et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626-34
113. Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073-8
114. Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N Engl J Med. 2008;359:1834-6
115. Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353:1652-4
116. Mitri Z, Constantine T, O'Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193
117. Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH. et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109-19
118. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25-40
119. Ploug M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr Pharm Des. 2003;9:1499-528
120. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23-36
121. Noh H, Hong S, Huang S. Role of urokinase receptor in tumor progression and development. Theranostics. 2013;3:487-95
122. Knor S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M. et al. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35:53-64
123. Jing YQ, Tong CL, Zhang J, Nakamura T, Iankov I, Russell SJ. et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69:1459-68
124. Lund IK, Illemann M, Thurison T, Christensen IJ, Hoyer-Hansen G. uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr Drug Targets. 2011;12:1744-60
125. Ngo JCK, Jiang LG, Lin ZH, Yuan C, Chen Z, Zhang X. et al. Structural basis for therapeutic intervention of uPA/uPAR system. Curr Drug Targets. 2011;12:1729-43
126. Jakobsche CE, McEnaney PJ, Zhang AX, Spiegel DA. Reprogramming urokinase into an antibody-recruiting anticancer agent. ACS Chem Biol. 2012;7:315-20
127. LeBeau AM, Duriseti S, Murphy ST, Pepin F, Hann B, Gray JW. et al. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013;73:2070-81
128. Wang M, Lowik D, Miller AD, Thanou M. Targeting the urokinase plasminogen activator receptor with synthetic self-assembly nanoparticles. Bioconjugate Chem. 2009;20:32-40
129. Yang L, Mao H, Cao ZH, Wang YA, Peng XH, Wang XX. et al. Molecular imaging of pancreatic cancer in an animal model using targeted multifunctional nanoparticles. Gastroenterology. 2009;136:1514-25
130. Abdalla MO, Karna P, Sajja HK, Mao H, Yates C, Turner T. et al. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149:314-22
131. Kriegbaum MC, Persson M, Haldager L, Alpizar-Alpizar W, Jacobsen B, Gardsvoll H. et al. Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes. Curr Drug Targets. 2011;12:1711-28
132. Persson M, Kjaer A. Urokinase-type plasminogen activator receptor (uPAR) as a promising new imaging target: potential clinical applications. Clin Physiol Funct Imaging. 2013;33:329-37
133. Ploug M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics. 2013;3:467-76
134. Persson M, Skovgaard D, Brandt-Larsen M, Christensen C, Madsen J, Nielsen CH. et al. First-in-human uPAR PET: imaging of cancer aggressiveness. Theranostics. 2015;5:1303-16
135. El-Sayed IH, Huang X, El-Sayed MA. Surface plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: applications in oral cancer. Nano Lett. 2005;5:829-34
136. Sun B, Ranganathan B, Feng S-S. Multifunctional poly (D, L-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials. 2008;29:475-86
137. Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112:335-40
138. Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656-9
139. Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M. et al. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano. 2013;7:2078-89
140. Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003;9:269-77
141. Zhukov NV, Tjulandin SA. Targeted therapy in the treatment of solid tumors: practice contradicts theory. Biochemistry (Mosc). 2008;73:605-18
142. Mayer RJ. Targeted therapy for advanced colorectal cancer-more is not always better. N Engl J Med. 2009;360:623-5
143. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3-26
144. Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337-41
145. Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009;20:708-14
146. Chitgupi U, Qin Y, Lovell JF. Targeted nanomaterials for phototherapy. Nanotheranostics. 2017;1:38-58
147. Li R, Zheng K, Hu P, Chen Z, Zhou S, Chen J. et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics. 2014;4:642-59
148. Peters T Jr. All about albumin: biochemistry, genetics, and medical applications. San Diego, USA Academic Press. 1996
149. Jeong H, Huh M, Lee SJ, Koo H, Kwon IC, Jeong SY. et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 2011;1:230-9
150. Schnitzer JE, Oh P. Antibodies to SPARC inhibit albumin binding to SPARC, gp60, and microvascular endothelium. Am J Physiol. 1992;263:H1872-9
151. Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132:171-83
152. Elsadek B, Kratz F. Impact of albumin on drug delivery-New applications on the horizon. J Control Release. 2012;157:4-28
153. Fanali G, di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P. Human serum albumin: from bench to bedside. Mol Asp Med. 2012;33:209-90
154. Chen JC, Chen NS, Huang JF, Wang JD, Huang MD. Derivatizable phthalocyanine with single carboxyl group: synthesis and purification. Inorg Chem Commun. 2006;9:313-5
155. Chen Z, Xu P, Chen J, Chen H, Hu P, Chen X. et al. Zinc phthalocyanine conjugated with the amino-terminal fragment of urokinase for tumor-targeting photodynamic therapy. Acta Biomater. 2014
156. Zhou X, Zheng K, Li R, Chen Z, Yuan C, Hu P. et al. A drug carrier targeting murine uPAR for photodynamic therapy and tumor imaging. Acta Biomater. 2015;23:116-26
157. Schleich N, Po C, Jacobs D, Ucakar B, Gallez B, Danhier F. et al. Comparison of active, passive and magnetic targeting to tumors of multifunctional paclitaxel/SPIO-loaded nanoparticles for tumor imaging and therapy. J Control Release. 2014;194:82-91
158. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8:5696-706
159. Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA. et al. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS Nano. 2010;4:5887-96
160. Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9:6551-9
161. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014
162. Bourzac K. News Feature: Cancer nanomedicine, reengineered. Proc Natl Acad Sci U S A. 2016;113:12600-3
163. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017;2:17024
164. Park S-m, Aalipour A, Vermesh O, Yu JH, Gambhir SS. Towards clinically translatable in vivo nanodiagnostics. Nat Rev Mater. 2017;2:17014
165. McNeil SE. Evaluation of nanomedicines: stick to the basics. Nat Rev Mater. 2016;1:16073
Author contact
![]() Corresponding author: Mingdong Huang, Email address: HMD_labedu.cn
Corresponding author: Mingdong Huang, Email address: HMD_labedu.cn