- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Physicochemical and imaging...
FMX pharmacokinetics and...
FMX pharmacokinetics and...
FMX imaging reveals tumor...
Late FMX imaging predominantly...
Effect of FMX on innate immune...
FMX modifications for enhanced...
Theranostic use of FMX
FMX reports enhanced TNP...
FMX predicts initial tumor...
Future directions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2017; 1(3):296-312. doi:10.7150/ntno.20564 This issue Cite
Review
Prediction of Anti-cancer Nanotherapy Efficacy by Imaging
Miles A. Miller1, 2, Sean Arlauckas1, Ralph Weissleder1, 2, 3 ![]()
1. Center for Systems Biology, Massachusetts General Hospital, USA;
2. Department of Radiology, Massachusetts General Hospital, USA;
3. Department of Systems Biology, Harvard Medical School, USA.
Received 2017-4-13; Accepted 2017-5-27; Published 2017-7-6
Abstract

Anticancer nanotherapeutics have shown mixed results in clinical trials, raising the questions of whether imaging should be used to i) identify patients with a higher likelihood of nanoparticle accumulation, ii) assess nanotherapeutic efficacy before traditional measures show response, and iii) guide adjuvant treatments to enhance therapeutic nanoparticle (TNP) delivery. Here we review the use of a clinically approved MRI nanoparticle (ferumoxytol, FMX) to predict TNP delivery and efficacy. It is becoming increasingly apparent that nanoparticles used for imaging, despite clearly distinct physicochemical properties, often co-localize with TNP in tumors. This evidence offers the possibility of using FMX as a generic “companion diagnostic” nanoparticle for multiple TNP formulations, thus potentially allowing many of the complex regulatory and cost challenges of other approaches to be avoided.
Keywords: dextran-coated iron oxide nanoparticle, magnetic resonance imaging, enhanced permeability and retention effect, nanomedicine, personalized medicine, tumor associated macrophage.
Introduction
Therapeutic nanoparticles (TNP) have been developed to deliver anticancer drugs more safely and effectively to various types of cancer. Many cytotoxic anticancer drugs exhibit poor pharmacokinetic profiles and low solubility, often requiring administration through prolonged intravenous infusion and the use of harmful solubilizing agents. The high peak concentrations of drug and solvent that result from intravenous injection can exacerbate systemic toxicities. To address this issue, drug encapsulation into nanoparticle (NP) carriers such as liposomes, micelles and polymeric NP, offers the possibilities of continuously releasing drug as the NP circulate over extended time periods and avoiding harsh solvents [1-5]. Decades of research have demonstrated the potential for therapeutic nano-encapsulation to improve drug safety while maintaining or even improving efficacy, leading to regulatory approval of several TNP for clinical use. Examples include nanoformulations of the DNA-damaging anthracycline drugs doxorubicin (Doxil, Myocet, Caelyx) and daunorubicin (DaunoXome); the microtubule-targeting drugs vincristine (Marqibo) and paclitaxel (Abraxane, Genexol-PM); the topoisomerase I inhibitor irinotecan (Onivyde); and the DNA-damaging antibiotic neocarzinostatin (SMANCS). Co-encapsulation of multiple drugs allows controlled treatment with synergistic drug combinations, exemplified by the liposomal 5:1 molar ratio formulation of cytarabine and daunorubicin (Vyxeos; Phase III trials completed). Beyond cytotoxic chemotherapy, nano-encapsulation has also been used to efficiently deliver immune-stimulating drugs, for example as with mifamurtide (Mepact) and its muramyl tripeptide payload that binds the pattern recognition receptor NOD2. Several more TNP are undergoing development in ongoing clinical trials [5, 6].
Although chemotherapy nano-encapsulation often reduces some of the side-effects observed with the un-encapsulated drug [3, 7], the impact on efficacy has been less clear in some cases [8-11]. Impressive efficacy has been achieved in recent trials for blood cancers such as acute myeloid leukemia [12], but findings in solid tumors have been more heterogeneous when comparing un-encapsulated and encapsulated formulations. This is especially problematic considering most FDA-approved TNP are indicated for solid tumors, including breast, ovarian, pancreatic, lung, and liver cancers. A meta-analysis of 14 clinical trials compared liposomal and conventional chemotherapy efficacy across >2,500 patients, finding no substantial difference in objective response, overall survival, and progression-free-survival [8]. This clinical experience contrasts with most studies using animal tumor models, where TNP superiority is typically reported [8, 13]. Consequently, these results raise the questions: what is driving heterogeneous patient response to TNP in solid cancers, and how can TNP clinical efficacy be better predicted?
Beyond merely improving drug safety, TNP are thought to improve efficacy through the enhanced delivery to solid tumors. Due to their size and other physicochemical properties, NP may passively accumulate in tumor tissue due to a variety of factors that are collectively known as the “enhanced permeability and retention” (EPR) effect [14, 15] or “tumor cell endocytosis” in earlier literature [16, 17]. In principle, disrupted and leaky tumor endothelium, high amounts of tumor-associated phagocytic immune cells, and dysfunctional lymphatic drainage all may contribute to the EPR effect that governs TNP delivery. Most data describing large EPR effects come from rapidly progressing mouse models of cancer, and particularly subcutaneous xenografts. Yet it is evident that many tumors in the clinic exhibit more complex tissue architecture and thus diverse EPR behavior. While some tumors may be highly perfused by abundant leaky vasculature, others exhibit substantial barriers to drug delivery including high interstitial fluid pressure, desmoplastic fibrotic encapsulation, and poor vascular perfusion (Fig. 1A). Several orthotopic and autochthonous tumor mouse models better reflect the latter clinical example, particularly for pancreatic cancer [18, 19], and have revealed how substantially TNP efficacy can be impeded by low EPR effects [20, 21]. Consequently, in patients, heterogeneous EPR effects across tumors may be a large factor in determining the delivery of TNP to tumors and the resulting impact on efficacy. The ability to observe the EPR effect in patients could thus be a useful tool for predicting how a patient will respond to TNP, and for determining whether a patient should receive TNP or an alternative treatment.
FMX imaging captures heterogeneous EPR effect in xenografts and patients. A) Schematic of solid tumors exhibiting varying degrees of EPR effect. B) Measurements of tumoral FMX accumulation were pooled from previously reported data across multiple xenograft mouse models (including orthotopic breast cancer) [22] and tumor lesions in patients [23]. Values were normalized to the average observed uptake in order to calculate coefficient of variation (CV = standard deviation / mean). Data are median ± interquartile range. C) Using quantitative FMX-MRI, concentrations of FMX in patient tumors were measured, in most cases across multiple metastatic lesions per patient. Patients were ordered according to median value for each tumor type (shown by bars). Figure is modified from [23].
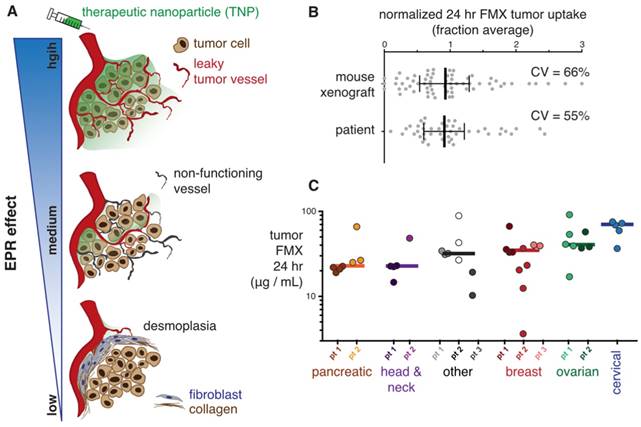
Clinical imaging potentially enables direct quantitation of the “EPR effect” when diagnostic NP are imaged. There are two general principles that have been pursued. First, one can label and image the TNP itself to observe its uptake into tumors. However, this is rarely done in clinical practice, because the regulatory and cost challenges can be substantial. Second, nano-sized imaging agents in clinical use may behave in a sufficiently similar manner to TNP so as to successfully enable prediction of TNP localization by imaging. Based on the hypothesis that the FDA-approved 30-nm magnetic NP, ferumoxytol (FMX, Feraheme), could predict co-localization of TNP by magnetic resonance imaging (MRI), we previously published that FMX-MR imaging correlated with uptake of TNP, cellular response, and inhibition of tumor growth in mouse xenograft models [22]. Subsequent work in the clinic has shown promising evidence of correlation between FMX imaging and tumor response [23]. FMX consequently may serve as a versatile complementary diagnostic for selecting patients to receive TNP or participate in TNP clinical trials.
This review focuses on the use of FMX-MRI as a surrogate to estimate TNP efficacy in vivo. We first focus on the biological properties of FMX, especially as observed using high-resolution in vivo confocal imaging (intravital microscopy) to understand the cellular basis of FMX-MRI enhancement. We then summarize key experiments that exemplify how FMX-MRI can be used for therapeutic purposes in the clinic.
Physicochemical and imaging properties of FMX
FMX is a product of evolution from earlier iron oxide NP, all of which exhibit superparamagnetic properties [24] and thus serve particularly well as T2-weighted MRI contrast agents. In the mid-1990's several first generation MR contrast agents were developed for imaging of the mononuclear phagocyte system (MPS; alternatively known as the reticulo-endothelial system, RES), including resident macrophages of the liver (Kupffer cells) [25]. A number of these agents were marketed worldwide under once-familiar names including Feridex® (Endorem®), Resovist® (Cliavist®), and Sinarem® (Combidex®). Feridex (Advanced Magnetics) had a relatively high polydispersity, large size (diameter 80-150 nm) and high magnetic relaxivity (R1 = 40 mM-1s-1; R2 = 160 mM-1s-1; 0.47T /37oC). Towards the end of the 2000's, manufacturers ceased commercial production of these agents due to lack of sales and clear clinical indications.
The addition of more extensive dextran coating to iron oxide NP enhances their in vivo stability and monodispersity. Compared to Feridex, these second generation monocrystalline iron oxide NP (MION), including Ferumoxtran (Combidex / Sinerem), maintained high relaxivity, were smaller (30-50 nm), more uniform and exhibited longer circulating half-lives (24 hr in humans and 11 hr in mice) [26]. With these improvements, imaging applications extended beyond the liver, for example facilitating the identification of lymph node metastases by their lack of particle uptake [27]. With additional refinements, MION have been synthesized with smaller size and higher relaxivity, as with MION-48 (26 nm diameter; R1 = 32.4 mM-1s-1; R2 = 130.5 mM-1s-1; 0.47T / 37oC). Dextran cross-linking, forming cross-linked iron oxide (CLIO; 29 nm diameter; R1 = 29 mM-1s-1; R2 = 74.3 mM-1s-1; 0.47T / 37oC), further stabilizes the NP and facilitates stable surface conjugation with labels and targeting ligands [28]. An early adaptation used annexin targeting to image apoptotic cells [29]. Ultrasmall superparamagnetic particles of iron oxide (USPIO) such as Resovist S (21 nm diameter; R1 = 24 mM-1s-1; R2 = 60 mM-1s-1) have also been developed for extrahepatic imaging [30]. Again, given challenges in markets and indications, several of these second generation NP were also withdrawn from the market (for example, Combidex, Resovist).
Ferumoxytol can be viewed as a 3rd generation iron oxide NP, and has been FDA-approved since 2009 for iron supplementation in patients with iron deficient anemia due to chronic kidney disease. FMX is widely available in the US (marked as Feraheme) along with Canada and Europe (marked as Rienso) [31, 32]. With a coating of semisynthetic, low molecular weight carboxymethyldextran, the reported particle size ranges between 17-31 nm in diameter, with relaxivities similar to other USPIO (R1 = 15 mM-1 s-1; R2 = 89 mM-1 s-1) [33]. FMX is generally well tolerated and is safer than the iron replacement therapy iron dextran (Dexiron / Infufer). At high 510 mg doses used for iron replacement therapy (roughly 5-10x the MR imaging dose of 1-2 mg / kg) it carries some risk of anaphylaxis, likely due to hypersensitivity to the carbohydrate, which prompted an FDA-mandated boxed warning on the package insert [34]. In clinical trials, serious hypersensitivity occurred in 0.2% of patients, with other less severe adverse reactions occurring in 2-4% [35]. However, in the post-approval setting, across roughly 80,000 chronic kidney disease patients over a 4 year period, risk of anaphylaxis was much lower at 0.03%. This risk was lower for FMX compared to iron dextran (0.08%) but higher than iron sucrose (0.02%) [36].
FMX's magnetic and biological properties support multiple MRI approaches. Negative-contrast (signal hypointensity) T2* imaging can be performed at field strengths above 0.5 T, with higher field strengths providing enhanced signal-to-noise. Furthermore, higher strengths increase transverse relaxation rates in surrounding tissue and allow shortened echo times, which reduces motion artifacts. Particularly at late time-points following FMX administration, T2 -weighted imaging has been useful for visualizing uptake by the MPS. As an example, FMX has been used to quantify infiltrating macrophages in atherosclerotic plaques [37] and myocardial infarctions [38]. Additionally, pathological lack of FMX uptake may appear as a positive signal against surrounding hypointense tissue. For instance, FMX has been used to stage disease by monitoring lack of FMX uptake in metastasis-bearing lymph nodes in prostate [39] and pancreatic [40] cancers. The relaxivity of iron oxide NP can dynamically respond to physicochemical changes to the NP and its environment, for instance, as it aggregates upon being internalized by cells [41]. This feature has been exploited in various applications, including the use of iron oxide NP to detect protein interactions [42]. Nevertheless, in the clinic, the primary imaging application is in monitoring FMX distribution and MPS uptake.
The ability of FMX to be safely administered relatively rapidly as a bolus i.v. injection allows it to be imaged as a blood pool contrast shortly after injection, for example as applied to stroke diagnosis [43]. Especially useful for blood pool measurements, signal-enhancement T1 imaging can be performed at standard clinical field strengths with short echo times [44]. Positive-contrast strategies under development involve exploiting off-resonance spin excitation [45, 46], suppression of on-resonance protons [47], gradient-compensation techniques, post-processing susceptibility-gradient mapping [48] and other techniques [49, 50].
Following systemic administration, the superparamagnetic properties of iron oxide nanoparticles change, as iron is bioavailable and is incorporated into the iron/heme pathway [25]. The temporal effects of SPIO on MRI signal have been quantitated [25] and as a general rule persist about 1-2 weeks in organs with highest uptake, namely the liver and spleen. After this time, the MR signal intensities of these organs return to normal.
For repeat imaging, such as for monitoring the evolution of inflammation [49], it is important to acquire matched pre- and post-FMX imaging, in order to correct for baseline changes due to residual FMX accumulation from prior doses. Importantly, prior FMX administration does not generally preclude the use of MRI for other applications such as follow-up diagnostics. Finally, if high therapeutic doses of FMX are given for anemia, package inserts caution that FMX can influence MRI for up to 3 months after its administration [35].
FMX pharmacokinetics and biodistribution in pre-clinical animal models
The pharmacokinetics and biodistribution of FMX have been extensively studied in various rodent models of disease. The initial blood half-life of FMX in rodents ranges between 1-2 hours depending on initial dose, and follows a similar distribution pattern to many other USPIOs [51, 52]. Because the blood half-life of FMX is an order of magnitude shorter in rodents than in humans, many pre-clinical studies adjust the dose of iron per mg body weight [53], leading to substantially-higher Cmax values that make comparisons to human pharmacokinetic models difficult. Tissue uptake in mice is predominantly in those with high levels of MPS activity. Thus, the spleen and liver are the predominant organs of FMX accumulation. Additional accumulation typically occurs at sites of inflammation such as in atherosclerotic plaques [54, 55], infarcted myocardium [56], along with xenograft and autochthonous tumors [16, 41, 57]. On a cellular level, FMX can be taken up in a variety of phagocytes including tissue-resident macrophages (e.g. Kupffer cells of the liver), circulating monocytes, and monocyte-derived macrophages at sites of inflammation and tumor tissue [58].
The transport and cellular uptake of fluorescently labeled FMX has been tracked at high resolution using in vivo (intravital) microscopy. Upon reaching tumor microvasculature, FMX is rapidly taken up into perivascular tumor-associated macrophages (TAM) that directly contact and in some cases extend cellular processes into the endothelium [22]. Over the course of 24 hr, FMX delivery is sustained by its long circulating half-life, and it gradually extravasates from vasculature into tumor tissue. Thus, TAM accumulation of FMX increases by 2-fold following the initial uptake period [22]. Although FMX is detectable in tumor cells themselves, its concentration in TAM is roughly 10-fold higher [22]. Intravital imaging of FMX has been performed in multiple tumor models, including orthotopic ovarian cancer, human fibrosarcoma [22], and syngeneic models of colon cancer [59]. In general, FMX accumulation is enhanced within the tumor compared to surrounding tissue, reflecting altered vascularization and phagocyte infiltration in many tumors [22].
Ex vivo immunofluorescence and histology analysis has confirmed FMX uptake primarily in TAM. Colocalization has been observed between the immunological macrophage markers F4/80 [22] and CD68 [53] in human fibrosarcoma xenograft and autochthonous breast cancer mouse models, respectively. FMX distribution has also been histologically characterized using an anti-dextran antibody to study brain tumor models in rats, again showing patterns of phagocyte-mediated FMX accumulation. In particular, FMX uptake was observed in astrocyte processes and endfeet surrounding tumor vessels, as well as in CD163(+)/CD68(+) macrophages [60]. As a type of glial cell, astrocytes are related to the microglia, which are the resident macrophages of the brain. In an orthotopic model of metastatic lung disease, fluorescently labeled CLIO was highly co-localized with macrophage markers CD68 and CD206 [61].
Complementary methods have further described which particular cell types accumulate FMX, and in what fraction of cells this occurs. For instance, comprehensive immunological profiling of FMX uptake has been performed using multicolor flow cytometry in an immunocompetent lung cancer model [22], confirming uptake into CD11b+ CD11c+ F4/80+ Ly6C- TAM [22]. Flow cytometry determined that the majority of tumor FMX uptake (70%) was due to TAM, with the remainder due to other phagocyte populations [22]. Tissue-clearing methods have been developed to image deep through entire tumors, enabling quantification of iron oxide NP uptake in TAM. Using a genetically engineered reporter mouse model bearing fluorescent macrophage populations (MERTKGFP/+), tissue clearing revealed that the majority (80%) of GFP+ TAM in an orthotopic lung cancer model accumulated CLIO, and >95% of NP+ cells were also GFP+ [61]. Results with FMX were similar: in a reporter mouse model expressing GFP+ macrophage and other myeloid cells (CX3CR1GFP/+), 90% of GFP+ cells in orthotopic lung tumors were FMX+, and >95% of FMX+ cells were GFP+ [22]. The efficiency with which fluorescently tagged FMX (and related USPIO) labels TAM has made it a valuable tool for studying the role of TAM in other applications. Examples include monitoring checkpoint immunotherapy [59], radiation therapy [62], and therapies targeting receptors such as colony-stimulating-factor 1 receptor (CSF1R) expressed on TAM [61].
In mouse models of cancer, tumoral FMX uptake can be highly heterogeneous. Even across xenograft tumors generated from the same cell line and implantation method, FMX-MRI shortly after FMX administration highlights the variability in tumor vascularization, with a coefficient of variation (CV) in ΔT2 of 65% [22]. FMX tumor concentration at later times post-administration (24 hr), which primarily reflects extravascular accumulation and TAM uptake, also exhibits high variability (Fig. 1B) [22]. Likewise, heterogeneity in the uptake of FMX and similar iron oxide NP has been reported in other orthotopic tumor models. In a brain tumor model in rats, baseline changes in cerebral brain volume over time, as measured by FMX-MRI, were similarly variable with CV of 50% [63]. Interestingly, tumors treated with the vascular modulating drugs intetumumab (αV-integrin targeting) and bevacizumab (vascular endothelial growth factor, VEGF, targeting) exhibited less variability (CVs of 15% and 30%, respectively) [63]. In a model of metastatic lung cancer, cellular uptake of iron oxide NP varied 8-fold across individual metastatic tumor nodules in lungs. The number of tumor-associated phagocytes that accumulated NP ranged from 6,000 - 80,000 per mm3 of tumor tissue, and this variability was even greater across tumors from different cell lines and lung-cancer types [61]. In this and other xenograft models, direct TAM modulation with therapies such as CSF1R-targeting inhibitors [53, 61] and clodronate liposome [22, 62] impacted FMX uptake and distribution within tumors. Overall, these data show that both tumor vascularization and phagocyte content contribute to the substantially observed heterogeneity across tumors and tumor-types.
FMX pharmacokinetics and biodistribution in humans
The ease with which FMX can be detected has provided a number of independent methods to model its pharmacokinetics in humans. The recommended therapeutic dose of FMX is 510 mg of elemental iron (roughly 6-8 mg/kg body weight) in 17 mL of isotonic solution containing mannitol, delivered intravenously at a rate of up to 30 mg sec-1. Blood half life increases with increasing dose, whereas the injection rate has no effect on the circulation time [64]. Despite its indication for chronic kidney disease, there is no difference in pharmacokinetic parameters in patients on hemodialysis, as FMX is not eliminated by renal filtration [64]. The semisynthetic carbohydrate coating permits long-circulation of the particle [44, 65]. This thick coating also reduces release of free iron into serum, which mitigates the clinical complications associated with alternative preparations of intravenous iron. Furthermore, this facilitates distinction of circulating particle from blood hemoglobin when performing pharmacokinetic assessments [66]. The blood half life is approximately 15 hrs (clearance CL = 69.1 mL / hr), and for a standard 510 mg dose, the max circulating concentration Cmax = 0.2 mg / mL is reached relatively quickly following infusion (tmax = 18 min). FMX has a low volume of distribution VD of 3.16 L, reflecting its initial confinement to the vasculature [35]. Iron is released from the colloidal particle after uptake and lysosomal degradation by macrophages in the liver, spleen, lymph nodes and bone marrow [67]. Radiotracer and MR relaxivity studies have shown how the iron-oxide NP core is broken down into heme [25]. While the dextran carbohydrate is excreted in the urine and feces, iron is retained and incorporated into the body's iron stores [68, 69].
Aside from its indication for iron supplementation, FMX has been successfully utilized in the clinic as an imaging agent. FMX-MRI enables selective enhancement in tumors, and may provide imaging-based delineation of tumor margins in pancreatic cancer [70]. Although most imaging of lymph node metastases has been performed with ferumoxtran [27], the approach has been extended to FMX [39, 40]. Doses as high as 7.5 mg / kg were found safe and useful for de-enhancing lymph nodes [71]. In the central nervous system, FMX-MRI better differentiated metastases from benign meningioma lesions in patients, compared to gadolinium-based contrast [72]. Immunopathology has been used to identify molecular correlates of FMX contrast enhancement. One study found FMX correlation with decreased expression of the cell-cell gap-junction marker Connexin-43, which suggested that FMX accumulation was related to increased penetration through permeable tissue [72]. In another report, FMX tumor imaging and histology showed uptake into vascularized, CD68+ macrophage-rich regions surrounding nests of tumor cells [23]. Given its tendency to accumulate in phagocytic immune cell populations, FMX-MRI has also been used to image inflammation outside of cancer. In application to diabetes, FMX-MRI signal-enhancement was attributed to macrophage uptake in inflamed pancreatic lesions [49].
As with the pre-clinical animal studies, FMX accumulation in patients is highly variable across different tumors and even tumor regions (Fig. 1C). FMX-MRI has shown greater diversity in various tumor vascularization features across patients compared to xenograft tumors [73]. High variability in FMX-MRI has also been observed in pancreatic adenocarcinoma (average CV 580% in T2 enhancement), which was attributed to mixed degrees of local inflammation and cancer cell / fibrosis ratios across tumors [70]. FMX-MRI variability has been likewise observed across metastases in individual patients (CV = 34%) and across different patients (CV = 55%) [23]. Although sample sizes were modest, FMX-MRI showed the lowest signal in pancreatic cancer lesions, which reflects substantial data describing low EPR effect in that cancer type [18-20, 74]. Across all lesions, MR enhancement varied by more than 8-fold. Interlesional variability within individual patients ranged from 1.03-fold to 4.22-fold. In other words, some patients exhibited highly similar FMX accumulation across all of their metastatic lesions, while others showed substantial heterogeneity. Variability was modest across different subparts of individual tumor lesions (average differences of 1.6-fold), although >10-fold variability was observed in some cases [23].
FMX imaging reveals tumor vascularization and permeability
Imaging FMX immediately upon systemic administration to patients reveals the extent and morphological characteristics of tumor vascularization, permitting quantification of vessel permeability (Fig. 2). Measurable features including vessel density, relative vascular volume fraction, and mean microvessel caliber or vessel size index have been imaged with FMX-MRI in tumors [49, 73]. Methods are being developed to use dual-contrast MRI for assessing vessel features combined with transvascular permeability. Dual-contrast MR images using iron oxide NP, ultrashort echo times pulse sequence, and conventional spin- and gradient-echo methods have enabled simultaneous quantitation of transvascular permeability (also known as the water exchange index), blood volume, and the vessel caliber index [75]. Dynamic susceptibility-weighted contrast-enhanced MRI (DSC-MRI) has been used with FMX to quantitate relative cerebral blood volume in brain cancer patients. Results show that FMX may be preferable to gadolinium-based contrast (gadoteridol) for imaging vasculature, since gadoteridol extravasates into tissue more rapidly than FMX, which has a much larger hydrodynamic radius by comparison. Early gadoteridol extravasation can make tumors erroneously appear as though they are progressing, and thus FMX may better distinguish tumor progression from pseudoprogression [76]. In relation to TNP delivery, clinical imaging studies have fit FMX-MRI data to compartmental pharmacokinetic models, and have found early signal enhancement highly correlates with vascular permeability in tumors [23] (Fig. 2A).
FMX imaging correlates with vessel permeability. A) Early FMX-MRI of tumors in patients was fit to a compartmental pharmacokinetic model, and correlation was observed between computationally inferred permeability (y-axis) and FMX (x-axis). The thick line and shading denote the fit and confidence interval of a linear regression model. Figure modified from [23]. B) Intravital microscopy in a xenograft mouse model was used to measure dynamic bursts of macromolecular extravasation, shown by rapid transport of dextran from vasculature into tumor tissue, as outlined by the orange dashed line. Scale bar = 20 μm. FMX was pre-injected 24 hr prior, and the timestamps denote time post-injection of the 60 kDa fluorescent dextran. C) Using data as in B, effective vessel permeability was measured for each image dataset and correlated with multiple other image features such as vessel width, cell density, and vessel branching. The enrichment of FMX+ tumor-associated phagocytes correlated most with permeability, suggesting these cells contribute to vessel leakiness. D) Bursts of dextran extravasation were identified by data as in B, and were found to occur more often in vessels with FMX+ phagocytes in close proximity. For B-D, figures are modified from [62].
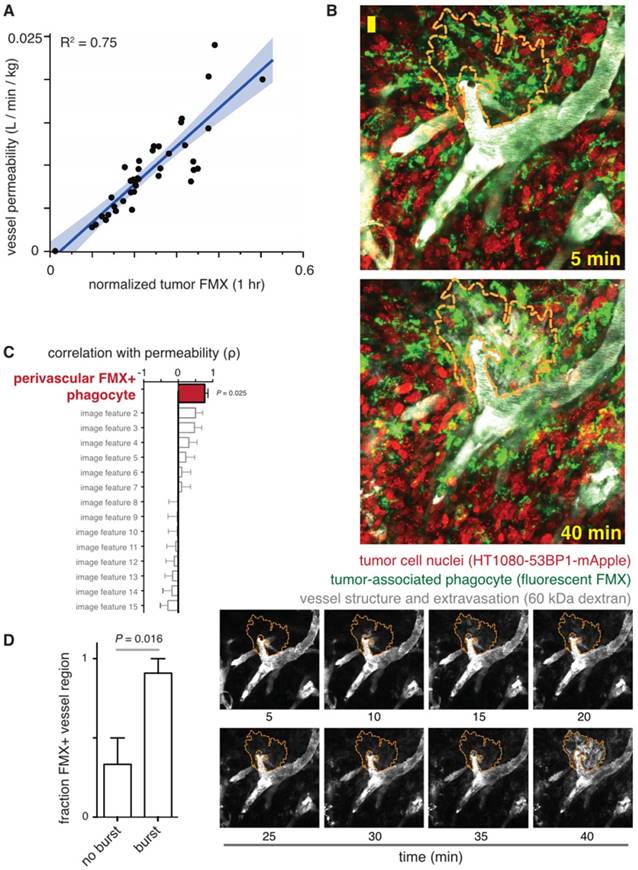
Even late FMX imaging (24 hr post-injection) may indirectly correlate with vessel features and permeability. At the cellular level, perivascular phagocytes accumulating FMX can interact with endothelium to influence permeability. For instance, reports have shown how perivascular TAM can produce angiogenic VEGF and induce transient endothelial permeability [77]. Intravital microscopy and experiments with VEGF inhibitor helped reveal how TAM-mediated vascular permeability was due to both VEGF signaling and mechanical disruption of endothelial integrity via cellular protrusion [77]. Similar perivascular phagocyte populations have been found to uptake high amounts of FMX, and their localization is correlated with bursts of macromolecular extravasation and TNP delivery [62] (Fig. 2B-D). TAM depletion [62] and VEGF inhibition [77] both reduce tumor vessel permeability in pre-clinical models.
Late FMX imaging predominantly captures tumor extracellular space and TAM uptake
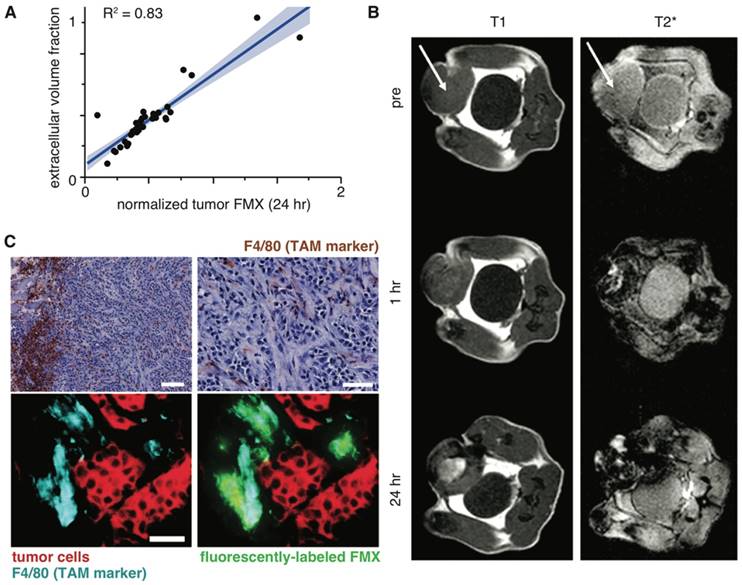
After FMX has cleared circulation, within tumors it primarily resides in extracellular space and within TAM. In patients, late FMX enhancement correlates strongly with extracellular volume fraction, as inferred using a compartmental pharmacokinetic model [23] (Fig. 3A). Similarly, computational analysis of intravital datasets has found the extracellular fraction within tumor tissue to be a major determinant of accumulation [22]. MRI methods have recently been developed to distinguish extracellular from intracellular FMX by comparing T1 and T2 enhancement ratios [78]. When applied to different mouse models of breast cancer, this method identified extracellular accumulation in the necrotic core of an implanted, orthotopic tumor model (Fig. 3B). In contrast, TAM uptake rather than extracellular accumulation was primarily observed in the autochthonous MMTV model [78]. In many cases, the combination of flow-cytometry, histology, and intravital microscopy have collectively shown FMX accumulates at the highest levels in TAM (Fig. 3C) [22, 53, 60, 61].
Late FMX uptake is driven by extracellular tissue space and TAM uptake. A) FMX-MRI measurements in patients were fit to a compartmental pharmacokinetic model, and correlation was observed between computationally inferred extracellular volume fraction (y-axis) and FMX (x-axis). Thick line and shading denote the fit and confidence interval of a linear regression model. Figure modified from [23]. B) T1-weighted and T2*-weighted FMX-MRI show positive and negative enhancement, respectively, upon FMX accumulation. In this example, T1-weighted images show extracellular FMX accumulation in the necrotic tumor core, while T2*-weighted images show intracellular uptake via phagocytes near the tumor periphery. Figure adapted from [78]. C) 24 hr post-injection with fluorescently-labeled FMX, fibrosarcoma tumors from a xenograft mouse model were excised and stained for hematoxylin and an F4/80 antibody at x10 (top left; scale bar 100 μm) and x40 (top right; scale bar 50 μm). Adjacent sections were imaged by immunofluorescence (bottom; x40, scale bar 50 μm). Reproduced from [22].
Effect of FMX on innate immune cells
FMX has filled a need in iron-deficient patients with chronic kidney disease, by supplementing nano-encapsulated iron to the MPS cells responsible for hemoglobin-recycling. The primary biological outcome of FMX administration is the elevation of hemoglobin levels [31]. Direct effects of iron on macrophage populations have been observed, and transition to a pro-inflammatory M1-type phenotype has been reported to impair wound healing via perpetuated TNFα and reactive oxygen species production [79]. Alternatively-activated macrophages shifted to a more classically-activated M1 phenotype in vitro when given an iron oxide NP, although investigations were not extended to animal studies [80]. Co-implantation of cancer cells suspended in high concentrations of FMX can lead to slower tumor growth in some models, and high-dose FMX pre-injection (10 mg Fe kg-1) can prevent metastatic growth in models of hematogenous metastasis [81]. FMX effects on tumor growth were macrophage-dependent, and pro-inflammatory macrophage expression signatures were observed under similar FMX treatment conditions [81]. These provocative findings are worth further investigating, especially to determine if they extend to humans and occur at the lower doses used for FMX imaging applications.
FMX modifications for enhanced functionality
Given their general safety and proven clinical utility, iron oxide NP show promise as platforms for the introduction of new drug delivery systems and multimodal imaging agents. For example, FMX can be modified to carry cytotoxic chemotherapeutic payloads such as doxorubicin, thus enabling non-invasive monitoring of drug delivery to tumors. The carbohydrate coating of FMX lends itself to hydrophobic drug loading based on electrostatic interactions. Moreover, changes in magnetic relaxation with drug association and pH-dependent in vivo release can be monitored by MRI [82]. Multiple anticancer drugs and combinations thereof have been efficiently loaded into FMX using this approach, including radiolabeled drugs that facilitate dual-modality imaging of the FMX vehicle by MRI and the therapeutic payload by PET [82].
Other methods have expanded beyond weak electrostatic interactions to conjugate PET ligands to FMX. For instance, FMX surface modification was used to chelate 89Zr, and this covalent attachment permits sensitive lymph node tracking for tumor staging and surgical resection [83]. Heat-induced radiolabeling (HIR) offers a non-covalent alternative for 89Zr conjugation, and is a less drastic modification of the surface chemistry with comparatively less impact on the pharmacokinetic profile of FMX [84]. Standard uptake value measurements with HIR-89Zr-FMX have provided one of the most sophisticated compartmental models of fast and slow uptake kinetics in mice, and flow cytometry of a fluorescently-labeled derivative helped attribute the slow uptake processes to monocyte trafficking [84]. A number of fluorescent dye-conjugated forms of FMX have been described to date, with applications ranging from flow cytometry, to microscopy, to in vivo imaging [22, 53, 69, 85, 86]. Chemically cross-linking the carbohydrate shell extends circulation time and has been widely used to achieve the biostability required for many imaging studies [87]. Further efforts to direct iron oxide NP to specific organs or diseased tissues require conjugation of targeting ligands to the particle surface, but selective in vivo targeting efficiency is often impeded or influenced by the layer of proteins that interact with NP in the blood, collectively referred to as the protein corona [88].
Theranostic use of FMX
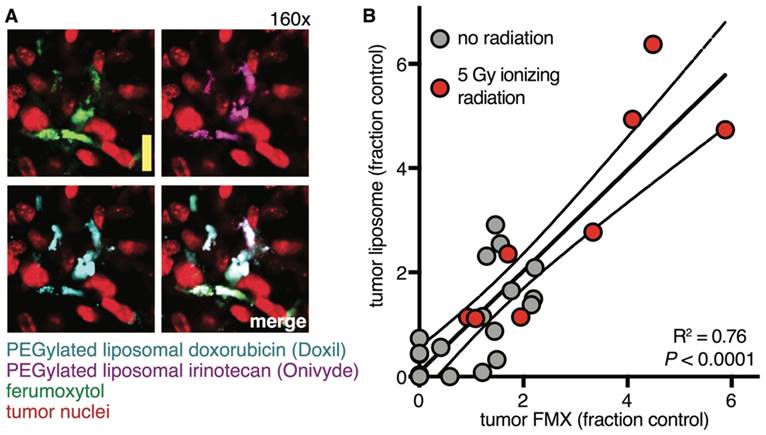
The physicochemical properties of FMX are distinct from typical TNP being tested in the clinic, such as those comprising micellar formulations of block co-polymers including poly(lactic co-glycolic) acid (PLGA) - b - polyethylene glycol, PLGA-PEG [9]. In general, polymeric micelles are larger than FMX. Rather than a carbohydrate dextran coating, polymeric micelles and many other TNP are coated in a PEG shell to reduce protein adsorption and immunogenicity. Polymer cores of TNP are also less stiff than the iron oxide core of FMX. Commonalities between the two NP include a particle size that exceeds the cutoff for renal excretion, comparable negative zeta-potential, and similar blood half-lives in mice [22]. PEGylated liposomal TNP formulations such as Doxil and Onivyde exhibit similar sizes and zeta-potentials compared to polymeric NP, but slightly longer blood half-lives in mouse xenograft models [62]. In humans, the 15 hr blood half-life of FMX is longer than the terminal half-life observed in some PLGA-PEG formulations (for instance, 6.5 hr [9]) but shorter than the observed 55 hr and 68 hr terminal half-lives observed for PEGylated liposomal doxorubicin (Doxil) and PEGylated liposomal irinotecan (Onivyde) [89, 90].
Intravital imaging reveals strong co-localization between FMX and TNP in tumor microvasculature, suggesting little discrimination in vessel perfusion between NP formulations. However, the kinetics of extravascular NP accumulation in tumor tissue is distinct. In vivo microscopy shows that while FMX accumulates rapidly in perivascular TAM at an apparently higher rate, the uptake of TNP and its extravasation into tissue is slower [22]. Nevertheless, at later time-points once the NP have cleared circulation, there is a high degree of overlap between FMX and TNP accumulation in TAM [22]. As evidence of this point, computational modeling has been used better predict TNP distributions by first quantitating tumor FMX, and then correcting for known kinetic differences between the NP to more accurately predict concentrations and kinetics of TNP accumulation in the same tissue [22]. Intravital imaging, flow cytometry, and histology collectively show 80-95% accuracy in the ability of FMX to label tumor-associated phagocytes that accumulate TNP [22, 62]. Especially with polymeric TNP formulations, uptake in tumor cells is greater than that observed with FMX. Nonetheless, accumulation of TNP in tumor cells is still roughly 30% less than in TAM [22]. Of note, these results describe TNP lacking tumor-specific molecular targeting ligands, which have been observed to further increase uptake in tumor cells rather than TAM [91].
Given their distinct sizes, one may expect there to be some differences in the accumulation of FMX and TNP across tumors. To address this question, past studies have directly compared tumoral distributions of differently-sized NP. One report found that when tumors were well vascularized and the vessels were permeable, both small and large NP co-accumulated at high amounts [20]. In contrast, evidence suggests that permeability-reducing therapeutics, such as anti-VEGF treatment, may reduce accumulation of larger NP more than smaller (< 40 nm) NP [92]. Differences in accumulation between contrasting NP may be greatest at microscopic (≤ 100 μm) length-scales, where patterns of subcellular localization [22] and tumor penetration may be distinct [93, 94].
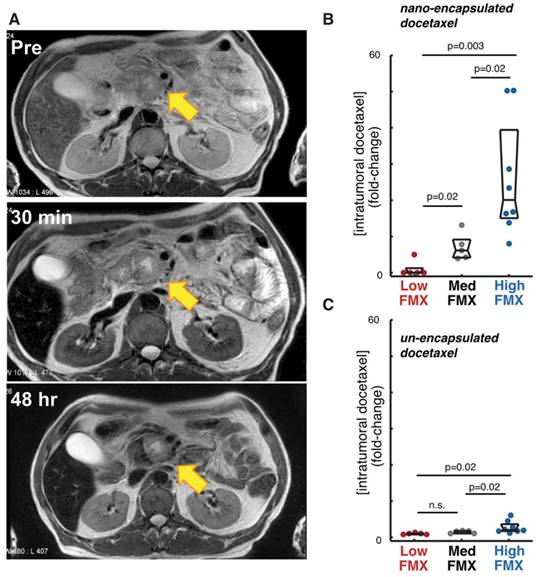
Nonetheless, multiple examples have been reported using various tumor models and NP, showing microscopic differences may be outweighed by general patterns of co-localization at mesoscopic/macroscopic length scales across the whole tumor [93, 94]. Indeed, late FMX imaging successfully correlated with accumulation of a model polymeric micelle TNP across a cohort of orthotopic breast cancer xenograft tumors (Fig. 4). FMX also correlated with accumulation of an analogous yet un-encapsulated formulation of the TNP payload, which likely reflects a general dependence on tumor vascularity and perfusion. Nonetheless, the magnitude of FMX correlation was much greater for the nano-formulation, suggesting a dominant influence of NP-specific “EPR effect”.
Correlation of FMX with drug uptake in tumors. A) FMX-MRI of a pancreatic cancer patient shows uptake and perfusion of the liver, kidneys, spleen, and subtle changes at the pancreas-tumor interface, with an apparent enrichment in TAM near the margin. Modified from [70]. B-C) FMX accumulation in a mouse model of breast cancer was used to stratify tumors into low, medium, and high FMX cohorts, which correlated more with accumulation of a co-administered fluorescent docetaxel TNP (B), compared to the un-encapsulated solvent formulation (C). Reproduced from [22].
FMX reports enhanced TNP delivery following tumor priming
Given the challenge of efficiently delivering TNP to tumors, growing attention has been placed on strategies to therapeutically alter, or “prime”, the tumor microenvironment to better accommodate TNP delivery. These strategies have been of particular focus for tumor-types like pancreatic cancer, which are associated with intrinsically low EPR effect and barriers to delivery such as desmoplasia and high interstitial fluid pressure. Pre-clinical studies have examined multiple strategies for tumor priming, including local radiation therapy [62, 95], cytotoxic chemotherapy [96], extracellular matrix-degrading treatments such as hyaluronidase [18], angiotensin-II receptor inhibitors such as losartan to affect vascular perfusion [19] or production of TAM [97], and TGFβ inhibitors to decrease fibrosis [98]. Acute treatments such as ultrasound, heat, magnetism, and hyperthermia are also being developed. However, a necessary component of these approaches will be to predictably and longitudinally monitor tumor remodeling in order to optimize strategies, assess efficacy, and guide the therapeutic course of the patient. Other studies have directly imaged TNP to monitor and guide tumor priming (for instance, [99]), demonstrating the concept in patients. Nonetheless, direct TNP imaging has been fairly limited in part due to cost and regulatory complexity.
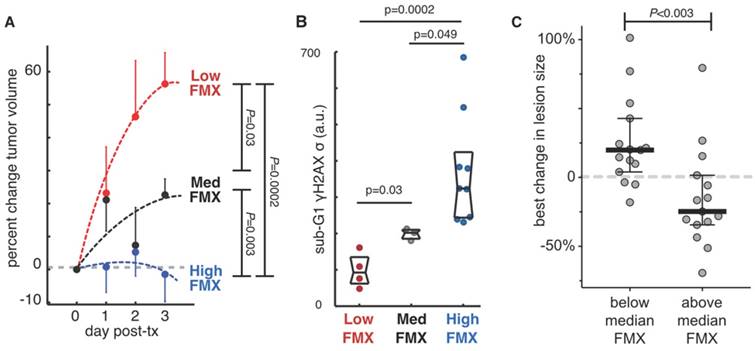
FMX-MRI has been successfully used to image patients undergoing neo-adjuvant chemoradiotherapy consisting of 25 Gy proton beam radiation and concurrent capecitabine. In this study, tumor priming enhanced FMX uptake, improved contrast enhancement, and suggested that tumors would be more likely to accumulate TNP [70]. In pre-clinical models, intravital microscopy revealed that neo-adjuvant radiation therapy and cyclophosphamide led to altered tumor vasculature, enhanced perivascular localization of TAM, and increased vessel permeability near FMX+ phagocytes [62]. Following tumor priming, FMX imaging across tumor cohorts showed correlation between FMX and accumulation of TNP (Fig. 5A). At the cellular level, radiation-primed tumors exhibited an enrichment in TAM, and nearly all TAM accumulating FMX had also accumulated various model TNP upon co-injection (Fig. 5B) [62]. In some cases, tumor priming was observed to increase TNP delivery by 6-fold [62]. Overall, these studies highlight the potential of FMX to predict not just baseline TNP delivery, but the success or failure of tumor priming to enhance TNP uptake.
FMX reports on enhanced TNP delivery following neo-adjuvant treatment. A) Fluorescently labeled versions of FMX and two FDA-approved TNP were co-injected into xenograft tumors that had been locally irradiated to enhance their EPR effects, and 24 hr later tumors were imaged by intravital microscopy (scale bar 20 μm). >90% of phagocytes accumulating one type of NP also accumulated the others. Reproduced from [62]. B) Fluorescently labeled FMX and a model liposome TNP were co-adminstered in tumor-bearing mice, and 24 hr later tumors were imaged for FMX and TNP accumulation. Some tumors were primed with local irradiation to enhance TNP delivery, which was reported by FMX. Each dot represents a tumor, while thick and dashed lines denote the fit and 95% confidence interval of a linear regression [62].
FMX predicts initial tumor response to TNP
Preclinical and clinical studies have now demonstrated the potential of FMX-MRI to predict not just the delivery of TNP, but its effect on tumor cell behavior and tumor growth. In a prospective translational experiment using a mouse xenograft model, a cohort of tumors was imaged by FMX-MRI, treated with a docetaxel-loaded TNP, and monitored for growth. After several days, tumors were excised and analyzed for cell cycle defects, DNA damage, and apoptosis. FMX-MRI results show that tumors with high enhancement at later time points were growth arrested, while tumors with low FMX-MRI enhancement grew fastest (Fig. 6A). These trends in tumor growth also correlated with aberrant cell cycle and DNA damage response (Fig. 6B) [22]. In the clinic, FMX-MRI correlated with patient response to TNP, as measured by best change in tumor burden (Fig. 6C) [23].
FMX predicts response to nanomedicine. A-B) FMX-MRI in a xenograft mouse model stratified tumors into low, medium, and high FMX cohorts. Following subsequent treatment with paclitaxel-encapsulated TNP, tumor growth was monitored for several days, revealing that tumor growth was only halted in tumors that accumulated high FMX (A). Tumor cell DNA damage and cell-cycle defects were measured by γH2A.X staining and flow cytometry, also correlating with FMX uptake (B). Reproduced from [22]. C) FMX-MRI measurements, performed across tumor lesions from 9 patients, were correlated with response to subsequently administered liposomal irinotecan TNP. Response was quantitated by changes in lesion size as imaged by CT at least 8 weeks after treatment initiation. Lesions with above-average FMX-MRI enhancement exhibited greater TNP response than lesions with below-average FMX accumulation. Modified from [23].
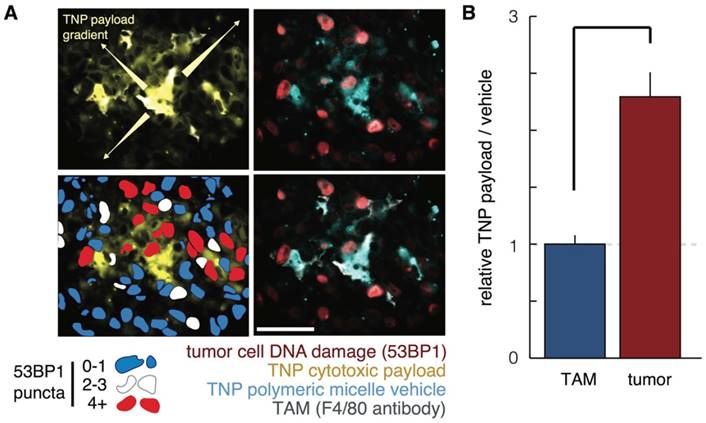
Several studies have shed light on the mechanistic basis for how FMX can predict TNP efficacy. Since FMX predicts TAM uptake of TNP, one question has been how TNP delivered to TAM in turn influence neighboring tumor cells. To answer this issue, multicolor intravital microscopy has been used to simultaneously image the TNP vehicle, the TNP payload, and DNA-damage response of individual cancer cells to treatment. Results show that cytotoxic TNP payload can slowly and locally redistribute from TAM to tumor cells in the immediate vicinity, leading to increased drug exposure and DNA damage in those cells (Fig. 7) [100]. In effect, TAM have the potential to serve as drug reservoirs that gradually deliver drug to tumor cells. With liposomal irinotecan, TAM have also been recognized as potentially mediating hydrolytic prodrug metabolism of irinotecan to its active form, SN-38 [101]. Drug redistribution from TAM to neighboring tumor cells partly explains the lower spatial co-localization between FMX and TNP payload, compared to FMX and TNP vehicle, that has been observed in pre-clinical models [22]. Likewise, spatial correlation at the microscopic level between FMX and TNP payload has been found to be modest in patients [23]. Despite evidence of spatial redistribution at the microscopic cellular level, FMX still effectively correlates with macroscopic readouts of tumor response in patients [23].
Mechanistic studies into how FMX predicts TNP efficacy. A-B) Polymeric micelle TNP were labeled with two fluorophores, such that the polymer vehicle and cytotoxic cisplatin-related payload could be simultaneously measured. A) Histological analysis of payload redistribution from TAM to tumor cells shows F4/80+ cells accumulate high levels of TNP, exhibit a local gradient of payload emanating from them, and tend to be neighboring tumor cells with high activity of the DNA damage response marker 53BP1. Scale bar = 50 μm. B) Using flow cytometry of excised xenograft tumors, cancer cells were observed to have more payload than TAM, relative to the corresponding amount of TNP vehicle in each cell. This data among others suggested payload had transferred from TAM to tumor cells. Reproduced from [100].
As discussed above, FMX also correlates with tumor vascularity and permeability, and thus FMX-MRI may also correlate with direct uptake of TNP into tumor cells. To parse these two TAM effects, dual-labeled TNP vehicle and therapeutic payload were analyzed following tumor priming with radiation therapy. For the polymeric micellar TNP that was tested, roughly half of the enhanced payload accumulation in tumor cells was due to direct TNP uptake, while the other half was the result of indirect delivery through initial TNP accumulation in TAM [62].
Future directions
Promising pre-clinical and initial clinical results have demonstrated the potential of FMX to predict TNP response, thus motivating its testing in other applications and broader trials. To date, work has primarily focused on polymeric micelle TNP [22], PEGylated liposomal irinotecan (Onivyde) [23], and the use of FMX in applications outside of TNP delivery. Nonetheless, intravital imaging studies suggest the approach may be applicable to other TNP that accumulate in TAM, including PEGylated liposomal doxorubicin (Doxil) [102] and others. Future work may also explore the ability of FMX to predict molecularly targeted TNP, such as doxorubicin-loaded HER2-targeted immunoliposome [96]. Many of the same factors controlling non-targeted (or so-called “passively” targeted) TNP also influence the behavior of targeted TNP. For instance, tumor priming with cyclophosphamide has been found to enhance tumoral accumulation of non-targeted [62] and targeted [96, 99] TNP alike. Thus, FMX may also be useful for predicting targeted TNP accumulation. Other therapies including oncolytic viruses, heavily PEGylated biologics, and nano-encapsulated nucleic-acid therapies including those based on RNA interference and CRISPR may similarly be governed by EPR effects and correlate with FMX-MRI. Additionally, there could be use in developing “correction factors” that account for measurable physicochemical and pharmacokinetic differences between FMX and various TNP, allowing FMX-MRI data to be appropriately adjusted during post-processing to predict a particular TNP of interest.
Features of vascular morphology that are quantified by early FMX-MRI may apply to predicting delivery and activity of drugs beyond TNP, including small molecules and antibodies. FMX-MRI has already demonstrated use in monitoring the vascular response to anti-angiogenic therapy [63, 103]. Because late FMX-MRI can predict TAM abundance, it could find application in predicting and monitoring the activity of TAM-targeted therapies including CSF1R inhibitors [104], anti-PD-L1 checkpoint blockade therapy [105], and antibody therapies that are influenced by Fcγ receptor activity on TAM [59]. Future work may further develop methods such as early FMX dual-contrast MRI, which assesses vessel features and permeability [75], combined with late FMX dual-contrast MRI, which can distinguish FMX in extracellular space from TAM uptake [78]. With optimization and standardization, such methods may find broader use in the clinic. In principle, combined early and late FMX-MRI can map quantifiable features of tumor vasculature (vessel density, relative vascular volume fraction, vessel size index, transvascular permeability) and tissue (TAM abundance, necrosis), respectively, that may inform choice of treatment and nano-formulation thereof for a patient, a cohort or an entire trial. Resulting data would have the potential to predict whether a patient should receive treatment with TNP or other types of therapies. For instance, high TAM abundance and vessel permeability would suggest the tumor may respond to TNP, while poor vascularization may indicate the need for a highly permeable small molecule therapeutic, or the use of neoadjuvant tumor priming to enhance TNP delivery.
FMX-MRI could logistically be incorporated into routine clinical care, and quantitative imaging readouts could be integrated with other diagnostic information such as histopathology and genomic mutational analysis to plan a course of treatment. In this regard, FMX-MRI would serve as a “complementary diagnostic” that is not required for the safe and effective use of a TNP, but aids in the benefit-risk decision making [106]. For this to happen, future clinical studies will need to further demonstrate the ability of FMX-MRI to predict the efficacy of multiple FDA-approved TNP. In the meantime, FMX-MRI could be developed as a companion diagnostic required for use of a given TNP that is still undergoing clinical trials, and could be used in the context of biomarker-guided adaptive trials already being implemented [107].
As discussed here, FMX-MRI has the most immediate applicability to solid tumors, especially those known to exhibit heterogeneous EPR effects, such as pancreatic cancer.
Outside of cancer, TNP are useful in delivering antibiotics, anti-inflammatories, and nucleic-acid based drugs to phagocytic cells at sites of inflammation and MPS clearance. As an example of the former, monocytes and macrophages have been targeted with nanomedicines in cardiovascular diseases, where they accumulate in atherosclerotic plaques [108]. Lesion vascularization and immune cell infiltration play a role in drug delivery and response in many other indications. For instance, several ongoing clinical trials are using FMX-MRI to monitor inflammation and vascular defects in the central nervous system [43, 109], and TNP are being developed to specifically target such pathologies [110].
The use of FMX as a companion diagnostic for TNP follows a broad trend in nanomedicine, where NP imaging is being used to scale dosage, model pharmacokinetics and predict treatment efficacy of TNP (Table 1) [111-113]. Radiographic imaging of endogenous tissue contrast can in some cases predict TNP behavior, as is the case with dynamic contrast enhanced computed tomography to visualize regions of EPR most prone to liposomal delivery [95]. In other cases, direct TNP labeling permits pharmacokinetics of the resultant “theranostic” to be measured [114, 115]. FMX is not the only NP that has been successfully used as a predictive companion diagnostic. For example, a companion PET imaging NP has predicted the accumulation and efficacy of TNP with distinct physicochemical properties, including liposomal and polymeric micelle formulations, in pre-clinical models [116]. Compared to FMX-MRI, PET imaging may be an attractive alternative for imaging indications where MRI interpretation is difficult. For breast cancer imaging, iodine liposomes as a mammography contrast have been used to predict DOXIL efficacy in a rat model [112]. While each approach has advantages and disadvantages, they collectively demonstrate the potential of an imaging NP to predict the accumulation and activity of a distinct TNP.
Clinical and pre-clinical TNP studies using imaging
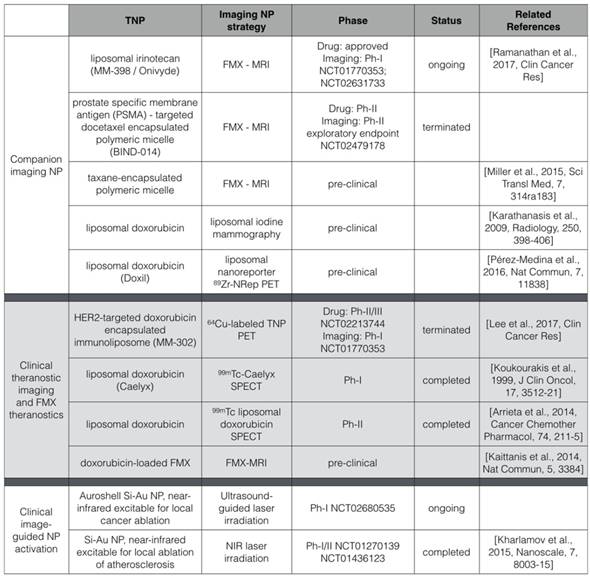
The mode by which TNP accumulate in tissues is complicated by many factors, including vascularity, lymphatic drainage, extracellular matrix composition, and immune cell infiltration, among others [117]. In tumor settings, the EPR effect has for years represented a simplistic view of the complicated interdependence between these factors [10, 118], but sophisticated new models are beginning to provide a better understanding NP pharmacokinetics [22, 23, 42, 62]. The ability to stratify patients based upon their likelihood to benefit from nanomedicine could improve the success rate of new TNP formulations, and this will rely on better predictive modeling of companion diagnostic - TNP combinations.
Abbreviations
CLIO: cross-linked iron oxide; CSF1R: colony stimulating factor 1 receptor; CT: computed tomography; CV: coefficient of variation; EPR: enhanced permeability and retention; FMX: ferumoxytol; GFP: green fluorescent protein; MION: monocrystalline iron oxide NP; MPS: mononuclear phagocyte system; MRI: magnetic resonance imaging; NP: nanoparticle; PET: positron emission tomography; RES: reticuloendothelial system; TAM: tumor-associated macrophage; TNP: therapeutic nanoparticle; USPIO: ultra-small particles of iron oxide; VEGF: vascular endothelial growth factor.
Acknowledgements
We thank Drs. Mukesh Harisinghani, Sandeep Hedgire and Rainer Kohler (MGH) for discussions and providing images.
Funding
This work was supported in parts by the NIH UO1CA206997, K99CA207744, R01EB010011 and P50GM107618.
Author contributions
M.A.M., S.A., and R.W. developed the concept and wrote the manuscript.
Data and materials availability
Requests for collaboration involving materials used in this research will be fulfilled provided that a written agreement is executed in advance between Massachusetts General Hospital and the requesting parties.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Juliano RL, Stamp D. Pharmacokinetics of liposome-encapsulated anti-tumor drugs. Studies with vinblastine, actinomycin D, cytosine arabinoside, and daunomycin. Biochem Pharmacol. 1978;27:21-7
2. Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283-318
3. O'Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A. et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440-9
4. Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108:1850-5
5. Min Y, Caster JM, Eblan MJ, Wang AZ. Clinical Translation of Nanomedicine. Chem Rev. 2015;115:11147-90
6. Sheridan C. Proof of concept for next-generation nanoparticle drugs in humans. Nat Biotechnol. 2012;30:471-3
7. Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P. et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794-803
8. Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J Control Release. 2016 S0168-3659:30241-3
9. Von Hoff DD, Mita MM, Ramanathan RK, Weiss GJ, Mita AC, LoRusso PM. et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22:3157-63
10. Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX. et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin Cancer Res. 2017 [Epub ahead of print]
11. Weiss GJ, Chao J, Neidhart JD, Ramanathan RK, Bassett D, Neidhart JA. et al. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Invest New Drugs. 2013;31:986-1000
12. Medeiros BC, Lancet JE, Cortes JE, Newell LF, Lin TL, Ritchie EK. et al. Analysis of Efficacy By Age for Patients Aged 60-75 with Untreated Secondary Acute Myeloid Leukemia (AML) Treated with CPX-351 Liposome Injection Versus Conventional Cytarabine and Daunorubicin in a Phase III Trial. Blood. 2016;128:902
13. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mat. 2016;16014:1-12
14. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
15. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271-84
16. Zimmer C, Weissleder R, Poss K, Bogdanova A, Wright SC, Enochs WS. MR imaging of phagocytosis in experimental gliomas. Radiology. 1995;197:533-8
17. Zimmer C, Wright SC, Engelhardt RT, Johnson GA, Kramm C, Breakefield XO. et al. Tumor cell endocytosis imaging facilitates delineation of the glioma-brain interface. Exp Neurol. 1997;143:61-9
18. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418-29
19. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV. et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516
20. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
21. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. 2015;219:192-204
22. Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH. et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7:314ra183
23. Ramanathan RK, Korn R, Raghunand N, Sachdev JC, Newbold RG, Jameson G. et al. Correlation Between Ferumoxytol Uptake in Tumor Lesions by MRI and Response to Nanoliposomal Irinotecan in Patients With Advanced Solid Tumors: A Pilot Study. Clin Cancer Res. 2017 [Epub ahead of print]
24. Weissleder R, Elizondo G, Wittenberg J, Rabito CA, Bengele HH, Josephson L. Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology. 1990;175:489-93
25. Weissleder R, Stark DD, Engelstad BL, Bacon BR, Compton CC, White DL. et al. Superparamagnetic iron oxide: pharmacokinetics and toxicity. AJR Am J Roentgenol. 1989;152:167-73
26. Shen T, Weissleder R, Papisov M, Bogdanov A, Brady TJ. Monocrystalline iron oxide nanocompounds (MION): physicochemical properties. Magn Reson Med. 1993;29:599-604
27. Harisinghani MG, Barentsz J, Hahn PF, Deserno WM, Tabatabaei S, van de Kaa CH. et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N Engl J Med. 2003;348:2491-9
28. Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;23:1418-23
29. Schellenberger EA, Sosnovik D, Weissleder R, Josephson L. Magneto/optical annexin V, a multimodal protein. Bioconjug Chem. 2004;15:1062-7
30. Settles M, Etzrodt M, Kosanke K, Schiemann M, Zimmermann A, Meier R. et al. Different capacity of monocyte subsets to phagocytose iron-oxide nanoparticles. PLoS One. 2011;6:e25197
31. Provenzano R, Schiller B, Rao M, Coyne D, Brenner L, Pereira BJ. Ferumoxytol as an intravenous iron replacement therapy in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:386-93
32. Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85:315-9
33. Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L. et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108:2064-110
34. Vasanawala SS, Nguyen KL, Hope MD, Bridges MD, Hope TA, Reeder SB. et al. Safety and technique of ferumoxytol administration for MRI. Magn Reson Med. 2016;75:2107-11
35. AMAG Pharmaceuticals L. Feraheme (ferumoxytol) Injection {package insert}. 2015.
36. Wang C, Graham DJ, Kane RC, Xie D, Wernecke M, Levenson M. et al. Comparative Risk of Anaphylactic Reactions Associated With Intravenous Iron Products. JAMA. 2015;314:2062-8
37. Morishige K, Kacher DF, Libby P, Josephson L, Ganz P, Weissleder R. et al. High-resolution magnetic resonance imaging enhanced with superparamagnetic nanoparticles measures macrophage burden in atherosclerosis. Circulation. 2010;122:1707-15
38. Yilmaz A, Dengler MA, van der Kuip H, Yildiz H, Rösch S, Klumpp S. et al. Imaging of myocardial infarction using ultrasmall superparamagnetic iron oxide nanoparticles: a human study using a multi-parametric cardiovascular magnetic resonance imaging approach. Eur Heart J. 2013;34:462-75
39. Harisinghani M, Ross RW, Guimaraes AR, Weissleder R. Utility of a new bolus-injectable nanoparticle for clinical cancer staging. Neoplasia. 2007;9:1160-5
40. McDermott S, Thayer SP, Fernandez-Del Castillo C, Mino-Kenudson M, Weissleder R, Harisinghani MG. Accurate prediction of nodal status in preoperative patients with pancreatic ductal adenocarcinoma using next-gen nanoparticle. Transl Oncol. 2013;6:670-5
41. Moore A, Weissleder R, Bogdanov A. Uptake of dextran-coated monocrystalline iron oxides in tumor cells and macrophages. J Magn Reson Imaging. 1997;7:1140-5
42. Perez JM, Josephson L, O'Loughlin T, Högemann D, Weissleder R. Magnetic relaxation switches capable of sensing molecular interactions. Nat Biotechnol. 2002;20:816-20
43. Dósa E, Tuladhar S, Muldoon LL, Hamilton BE, Rooney WD, Neuwelt EA. MRI using ferumoxytol improves the visualization of central nervous system vascular malformations. Stroke. 2011;42:1581-8
44. Li W, Tutton S, Vu AT, Pierchala L, Li BS, Lewis JM. et al. First-pass contrast-enhanced magnetic resonance angiography in humans using ferumoxytol, a novel ultrasmall superparamagnetic iron oxide (USPIO)-based blood pool agent. J Magn Reson Imaging. 2005;21:46-52
45. Cunningham CH, Arai T, Yang PC, McConnell MV, Pauly JM, Conolly SM. Positive contrast magnetic resonance imaging of cells labeled with magnetic nanoparticles. Magn Reson Med. 2005;53:999-1005
46. Mani V, Briley-Saebo KC, Itskovich VV, Samber DD, Fayad ZA. Gradient echo acquisition for superparamagnetic particles with positive contrast (GRASP): sequence characterization in membrane and glass superparamagnetic iron oxide phantoms at 1.5T and 3T. Magn Reson Med. 2006;55:126-35
47. Stuber M, Gilson WD, Schaer M, Bulte JW, Kraichman DL. Shedding light on the dark spot with IRON—a method that generates positive contrast in the presence of superparamagnetic nanoparticles. Proc Intl Soc Magn Reson Med. 2005;13:2608
48. Liu W, Dahnke H, Jordan EK, Schaeffter T, Frank JA. In vivo MRI using positive-contrast techniques in detection of cells labeled with superparamagnetic iron oxide nanoparticles. NMR Biomed. 2008;21:242-50
49. Gaglia JL, Harisinghani M, Aganj I, Wojtkiewicz GR, Hedgire S, Benoist C. et al. Noninvasive mapping of pancreatic inflammation in recent-onset type-1 diabetes patients. Proc Natl Acad Sci U S A. 2015;112:2139-44
50. Hedgire SS, McDermott S, Wojtkiewicz GR, Abtahi SM, Harisinghani M, Gaglia JL. Evaluation of renal quantitative T2* changes on MRI following administration of ferumoxytol as a T2* contrast agent. Int J Nanomedicine. 2014;9:2101-7
51. Simon GH, von Vopelius-Feldt J, Fu Y, Schlegel J, Pinotek G, Wendland MF. et al. Ultrasmall supraparamagnetic iron oxide-enhanced magnetic resonance imaging of antigen-induced arthritis: a comparative study between SHU 555 C, ferumoxtran-10, and ferumoxytol. Invest Radiol. 2006;41:45-51
52. Leimgruber A, Berger C, Cortez-Retamozo V, Etzrodt M, Newton AP, Waterman P. et al. Behavior of endogenous tumor-associated macrophages assessed in vivo using a functionalized nanoparticle. Neoplasia. 2009;11:459-68
53. Daldrup-Link HE, Golovko D, Ruffell B, Denardo DG, Castaneda R, Ansari C. et al. MRI of tumor-associated macrophages with clinically applicable iron oxide nanoparticles. Clin Cancer Res. 2011;17:5695-704
54. Hyafil F, Laissy JP, Mazighi M, Tchétché D, Louedec L, Adle-Biassette H. et al. Ferumoxtran-10-enhanced MRI of the hypercholesterolemic rabbit aorta: relationship between signal loss and macrophage infiltration. Arterioscler Thromb Vasc Biol. 2006;26:176-81
55. Trivedi RA, Mallawarachi C, U-King-Im JM, Graves MJ, Horsley J, Goddard MJ. et al. Identifying inflamed carotid plaques using in vivo USPIO-enhanced MR imaging to label plaque macrophages. Arterioscler Thromb Vasc Biol. 2006;26:1601-6
56. Montet-Abou K, Daire JL, Hyacinthe JN, Jorge-Costa M, Grosdemange K, Mach F. et al. In vivo labelling of resting monocytes in the reticuloendothelial system with fluorescent iron oxide nanoparticles prior to injury reveals that they are mobilized to infarcted myocardium. Eur Heart J. 2010;31:1410-20
57. Turetschek K, Roberts TP, Floyd E, Preda A, Novikov V, Shames DM. et al. Tumor microvascular characterization using ultrasmall superparamagnetic iron oxide particles (USPIO) in an experimental breast cancer model. J Magn Reson Imaging. 2001;13:882-8
58. Kalber TL, Smith CJ, Howe FA, Griffiths JR, Ryan AJ, Waterton JC. et al. A longitudinal study of R2* and R2 magnetic resonance imaging relaxation rate measurements in murine liver after a single administration of 3 different iron oxide-based contrast agents. Invest Radiol. 2005;40:784-91
59. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS. et al. In vivo imaging reveals a tumor-associated macrophage mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017;9:eaal3604
60. McConnell HL, Schwartz DL, Richardson BE, Woltjer RL, Muldoon LL, Neuwelt EA. Ferumoxytol nanoparticle uptake in brain during acute neuroinflammation is cell-specific. Nanomedicine. 2016;12:1535-42
61. Cuccarese MF, Dubach JM, Pfirschke C, Engblom C, Garris C, Miller MA. et al. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat Commun. 2017;8:14293
62. Miller MA, Chandra R, Cuccarese MF, Pfirschke C, Engblom C, Stapleton S. et al. Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci Transl Med. 2017;9:eaal0225
63. Muldoon LL, Gahramanov S, Li X, Marshall DJ, Kraemer DF, Neuwelt EA. Dynamic magnetic resonance imaging assessment of vascular targeting agent effects in rat intracerebral tumor models. Neuro Oncol. 2011;13:51-60
64. Landry R, Jacobs PM, Davis R, Shenouda M, Bolton WK. Pharmacokinetic study of ferumoxytol: a new iron replacement therapy in normal subjects and hemodialysis patients. Am J Nephrol. 2005;25:400-10
65. Ersoy H, Jacobs P, Kent CK, Prince MR. Blood pool MR angiography of aortic stent-graft endoleak. AJR Am J Roentgenol. 2004;182:1181-6
66. Balakrishnan VS, Rao M, Kausz AT, Brenner L, Pereira BJ, Frigo TB. et al. Physicochemical properties of ferumoxytol, a new intravenous iron preparation. Eur J Clin Invest. 2009;39:489-96
67. Bashir MR, Bhatti L, Marin D, Nelson RC. Emerging applications for ferumoxytol as a contrast agent in MRI. J Magn Reson Imaging. 2015;41:884-98
68. Storey P, Lim RP, Chandarana H, Rosenkrantz AB, Kim D, Stoffel DR. et al. MRI assessment of hepatic iron clearance rates after USPIO administration in healthy adults. Invest Radiol. 2012;47:717-24
69. Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13:125-38
70. Hedgire SS, Mino-Kenudson M, Elmi A, Thayer S, Fernandez-del Castillo C, Harisinghani MG. Enhanced primary tumor delineation in pancreatic adenocarcinoma using ultrasmall super paramagnetic iron oxide nanoparticle-ferumoxytol: an initial experience with histopathologic correlation. Int J Nanomedicine. 2014;9:1891-6
71. Turkbey B, Agarwal HK, Shih J, Bernardo M, McKinney YL, Daar D. et al. A Phase I Dosing Study of Ferumoxytol for MR Lymphography at 3 T in Patients With Prostate Cancer. AJR Am J Roentgenol. 2015;205:64-9
72. Hamilton BE, Woltjer RL, Prola-Netto J, Nesbit GM, Gahramanov S, Pham T. et al. Ferumoxytol-enhanced MRI differentiation of meningioma from dural metastases: a pilot study with immunohistochemical observations. J Neurooncol. 2016;129:301-9
73. Fredrickson J, Serkova NJ, Wyatt SK, Carano RA, Pirzkall A, Rhee I. et al. Clinical translation of ferumoxytol-based vessel size imaging (VSI): miFeasibility in a phase I oncology clinical trial population. Magn Reson Med. 2017;77:814-25
74. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735-47
75. Kwon HJ, Shim WH, Cho G, Cho HJ, Jung HS, Lee CK. et al. Simultaneous evaluation of vascular morphology, blood volume and transvascular permeability using SPION-based, dual-contrast MRI: imaging optimization and feasibility test. NMR Biomed. 2015;28:624-32
76. Gahramanov S, Raslan AM, Muldoon LL, Hamilton BE, Rooney WD, Varallyay CG. et al. Potential for differentiation of pseudoprogression from true tumor progression with dynamic susceptibility-weighted contrast-enhanced magnetic resonance imaging using ferumoxytol vs. gadoteridol: a pilot study. Int J Radiat Oncol Biol Phys. 2011;79:514-23
77. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ. et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015;5:932-43
78. Aghighi M, Golovko D, Ansari C, Marina NM, Pisani L, Kurlander L. et al. Imaging Tumor Necrosis with Ferumoxytol. PLoS One. 2015;10:e0142665
79. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H. et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985-97
80. Laskar A, Eilertsen J, Li W, Yuan XM. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochem Biophys Res Commun. 2013;441:737-42
81. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
82. Kaittanis C, Shaffer TM, Ogirala A, Santra S, Perez JM, Chiosis G. et al. Environment-responsive nanophores for therapy and treatment monitoring via molecular MRI quenching. Nat Commun. 2014;5:3384
83. Thorek DL, Ulmert D, Diop NF, Lupu ME, Doran MG, Huang R. et al. Non-invasive mapping of deep-tissue lymph nodes in live animals using a multimodal PET/MRI nanoparticle. Nat Commun. 2014;5:3097
84. Normandin MD, Yuan H, Wilks MQ, Chen HH, Kinsella JM, Cho H. et al. Heat-Induced Radiolabeling of Nanoparticles for Monocyte Tracking by PET. Angew Chem Int Ed Engl. 2015;54:13002-6
85. Habibollahi P, Waldron T, Heidari P, Cho HS, Alcantara D, Josephson L. et al. Fluorescent nanoparticle imaging allows noninvasive evaluation of immune cell modulation in esophageal dysplasia. Mol Imaging. 2014;13:1-11
86. Yuan H, Wilks MQ, El Fakhri G, Normandin MD, Kaittanis C, Josephson L. Heat-induced-radiolabeling and click chemistry: A powerful combination for generating multifunctional nanomaterials. PLoS One. 2017;12:e0172722
87. Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc Chem Res. 2011;44:842-52
88. Bakhtiary Z, Saei AA, Hajipour MJ, Raoufi M, Vermesh O, Mahmoudi M. Targeted superparamagnetic iron oxide nanoparticles for early detection of cancer: Possibilities and challenges. Nanomedicine. 2016;12:287-307
89. Merrimack Pharmaceuticals. ONIVYDE (irinotecan liposome injection) {package insert}. 2010.
90. Janssen Products. Doxil (doxorubicin HCl liposome injection) {package insert}. 2007.
91. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB. et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732-40
92. Jiang W, Huang Y, An Y, Kim BY. Remodeling Tumor Vasculature to Enhance Delivery of Intermediate-Sized Nanoparticles. ACS Nano. 2015;9:8689-96
93. Shukla S, Ablack AL, Wen AM, Lee KL, Lewis JD, Steinmetz NF. Increased tumor homing and tissue penetration of the filamentous plant viral nanoparticle Potato virus X. Mol Pharm. 2013;10:33-42
94. Roode LE, Brighton H, Bo T, Perry JL, Parrott MC, Kersey F. et al. Subtumoral analysis of PRINT nanoparticle distribution reveals targeting variation based on cellular and particle properties. Nanomedicine (Lond). 2016;12:1053-62
95. Stapleton S, Allen C, Pintilie M, Jaffray DA. Tumor perfusion imaging predicts the intra-tumoral accumulation of liposomes. J Control Release. 2013;172:351-7
96. Geretti E, Leonard SC, Dumont N, Lee H, Zheng J, De Souza R. et al. Cyclophosphamide-Mediated Tumor Priming for Enhanced Delivery and Antitumor Activity of HER2-Targeted Liposomal Doxorubicin (MM-302). Mol Cancer Ther. 2015;14:2060-71
97. Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW. et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38:296-308
98. Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K. et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2012;109:16618-23
99. Miller K, LoRusso PM, Munster P, Ma C, Lee H, Reynolds J. et al. Effect of pre-treatment with cyclophosphamide on MM-302 (HER2-targeted liposomal doxorubicin) deposition in HER2-positive metastatic breast cancer patients assessed by 64Cu-MM-302 PET/CT. Cancer Res. 2015;75:P4-15
100. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C. et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. 2015;6:8692
101. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271-7
102. Seynhaeve AL, Dicheva BM, Hoving S, Koning GA, ten Hagen TL. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: evaluated by in vitro/in vivo live cell imaging. J Control Release. 2013;172:330-40
103. Pope WB, Young JR, Ellingson BM. Advances in MRI assessment of gliomas and response to anti-VEGF therapy. Curr Neurol Neurosci Rep. 2011;11:336-44
104. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264-72
105. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH. et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064-74
106. Scheerens H, Malong A, Bassett K, Boyd Z, Gupta V, Harris J. et al. Current Status of Companion and Complementary Diagnostics: Strategic Considerations for Development and Launch. Clin Transl Sci. 2017;10:84-92
107. Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Tsao A. et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44-53
108. Lameijer MA, Tang J, Nahrendorf M, Beelen RH, Mulder WJ. Monocytes and macrophages as nanomedicinal targets for improved diagnosis and treatment of disease. Expert Rev Mol Diagn. 2013;13:567-80
109. Hasan DM, Amans M, Tihan T, Hess C, Guo Y, Cha S. et al. Ferumoxytol-enhanced MRI to Image Inflammation within Human Brain Arteriovenous Malformations: A Pilot Investigation. Transl Stroke Res. 2012;3:166-73
110. Lee HJ, Park J, Yoon OJ, Kim HW, Lee DY, Kim DH. et al. Amine-modified single-walled carbon nanotubes protect neurons from injury in a rat stroke model. Nat Nanotechnol. 2011;6:121-5
111. Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG. et al. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res. 2001;7:243-54
112. Karathanasis E, Suryanarayanan S, Balusu SR, McNeeley K, Sechopoulos I, Karellas A. et al. Imaging nanoprobe for prediction of outcome of nanoparticle chemotherapy by using mammography. Radiology. 2009;250:398-406
113. Zheng J, Jaffray D, Allen C. Quantitative CT imaging of the spatial and temporal distribution of liposomes in a rabbit tumor model. Mol Pharm. 2009;6:571-80
114. Koukourakis MI, Koukouraki S, Giatromanolaki A, Archimandritis SC, Skarlatos J, Beroukas K. et al. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer. J Clin Oncol. 1999;17:3512-21
115. Arrieta O, Medina LA, Estrada-Lobato E, Ramírez-Tirado LA, Mendoza-García VO, de la Garza-Salazar J. High liposomal doxorubicin tumour tissue distribution, as determined by radiopharmaceutical labelling with (99m)Tc-LD, is associated with the response and survival of patients with unresectable pleural mesothelioma treated with a combination of liposomal doxorubicin and cisplatin. Cancer Chemother Pharmacol. 2014;74:211-5
116. Pérez-Medina C, Abdel-Atti D, Tang J, Zhao Y, Fayad ZA, Lewis JS. et al. Nanoreporter PET predicts the efficacy of anti-cancer nanotherapy. Nat Commun. 2016;7:11838
117. Torosean S, Flynn B, Axelsson J, Gunn J, Samkoe KS, Hasan T. et al. Nanoparticle uptake in tumors is mediated by the interplay of vascular and collagen density with interstitial pressure. Nanomedicine. 2013;9:151-8
118. Gabizon AA. Selective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomes. Cancer Res. 1992;52:891-6
Author contact
![]() Corresponding author: rweisslederharvard.edu
Corresponding author: rweisslederharvard.edu